Genomu Místě
Cesta analýza se stal takový společný postup v bioinformatice, zejména při studiu genové exprese. Pokud se podíváte na průzkum nedávných dokumentů, zdá se, že existuje spousta způsobů, jak to lze udělat. V tomto příspěvku budu diskutovat o rozdílech mezi dvěma běžně používanými nástroji; DAVID a GSEA.

koncepty za dva algoritmy jsou velmi odlišné., DAVID určuje překrývání mezi uživatelem dodaný gen seznamy a kurátor databází, hledat přesahy, které jsou větší, než se očekávalo náhodou. Přesnost algoritmu můžete zlepšit poskytnutím souboru na pozadí, který obsahuje všechny geny, které byly v experimentu považovány/detekovány. Genové sady dodávané uživatelem jsou obvykle generovány výběrem genů, které procházejí prahem významnosti. Postup DAVID je podobný ostatním dostupným, jako je vynalézavost, AmiGO a GeneGO., Výběr hodnot významnosti je do značné míry libovolný, ale je běžné nastavit prahovou hodnotu na FDR upravenou p<0.05. Změna prahu výrazně změní výsledky obohacení. Mezi použité statistické testy patří hypergeometrická, Fisherova přesná a Chi-čtvercová.
na rozdíl od výše uvedených metod, které vyžadují seznamy genů, které projdou svévolné limity, GSEA je nástroj, který používá každý datapoint ve svých statistických algoritmus. V“ klasické “ metodě jsou geny řazeny od většiny up-regulovaných až po většinu dolů regulovaných., Samotná metrika hodnosti se liší, ale dvě platné metody mají používat podepsanou hodnotu p nebo nižší interval spolehlivosti 90% změny záhybu. Základem testu je posoudit, zda členové gen se zobrazí obohacený na jednom konci profilu. Pro testování obohacení provádí GSEA permutace profilu a vypočítává obohacení genu tisíckrát nebo vícekrát, aby empiricky odhadl hodnoty p. Další nástroje, které používají celý profil, provádějí analytické testy, jako je test Wilcoxon a Kolmogorov-Smirnov.,
takže teď, když chápeme hlavní rozdíly v algoritmech, můžeme začít porovnávat výsledky z těchto dvou technik. Jsem se vrátil dataset probral jsem před léčbou azacitidinem v AML3 buňky profilované RNA-seq. Použiji diferenciální expresní data RNA-seq generovaná DESeq jako výchozí bod pro porovnání Davida a GSEA.
pro Davida jsem vytvořil soubor na pozadí sestávající ze všech 19 030 genů detekovaných v experimentu nad prahem detekce. Významný (FDR< 0.,05) seznamy genů ve směru nahoru (2502 genů) a dolů (2907 genů) byly odděleně předloženy Davidovi 6.7 pro analýzu se zaměřením pouze na keggovy cesty.
Pro GSEA jsem použil stejný metrický postup, který byl popsán dříve. Použil jsem klasický algoritmus GSEA s gsea2-2.2.2.jar spustitelný soubor, pomocí genových sad KEGG z MSigDB V5. 1.
v níže uvedeném grafu jsem určil překrytí statisticky významných genových sad (FDR<0.05) odděleně pro regulované dráhy nahoru a dolů., Také jsem zahrnul průsečík Davidových výsledků, když není poskytnuto žádné pozadí (pouze pro zvědavost, nikoli doporučený postup).
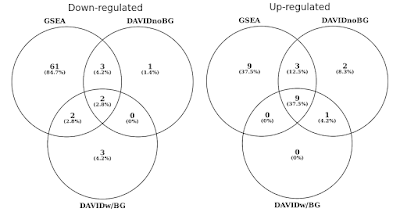
DAVID vs GSEA pomocí stejného profilu genové exprese. Čísla představují genové sady, které jsou významně obohaceny (FDR<0.05).
všimnete si, že v regulovaném směru nahoru i dolů identifikuje GSEA významnější výsledky ve srovnání s Davidem. Zjistil jsem, že to byl také případ v jiných knihovnách genů, jako je REACTOME a GO., Rozdíl v citlivosti mezi oběma nástroji se scvrkává na různé algoritmy, s Davidem pomocí diskrétního seznamu genů, versus GSEA, který používá každý datový bod. GSEA bude mnohem citlivější při identifikaci genových sad, které mají mnoho členů, kteří procházejí jemnými změnami v expresi. Individuálně, tyto změny nejsou statisticky významné, ale když velká část genů v sadě společně posun v projevu, že trend by mohl být velmi významný, statisticky vzato.,
kromě citlivějšího algoritmu jsou Davidovy genové sady a anotace genů opravdu zastaralé. Poslední aktualizace Davidových mnoha zdrojů dat je v polovině až do konce roku 2009. Jako takový, mnoho genových přístupů z Ensembl nemusí být podporováno. Vskutku, z mého souboru pozadí, který obsahoval 19,030 pouze 13,488 byl uznán Davidem. Nedostatek aktualizovaných genových sad je také problémem, stejně jako stále rostoucí počet citací, které DAVID stále přijímá.,
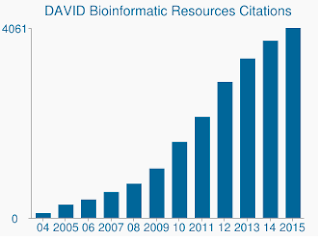
DAVID stále dostává tisíce citací ročně, přestože základní data jsou žalostně zastaralá.
Na závěr bych doporučil výzkumníci používají nástroje, které podívejte se na celý profil určit obohacení (GSEA, KAMERA, WilcoxGST, Pathifier), spíše než předložení genových sad pro analýzy překrytí. Použití Davida může být rychlá a snadná volba, ale to přichází na úkor masivního snížení citlivosti.
Další čtení
JA Timmons, KJ Szkop, IJ Gallagher., Více zdrojů zkreslení zmást funkční obohacení analýzy globálně-omics dat. Genomová biologie, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Dopad akumulace znalostí na analýzu obohacování dráhy. bioRxiv. doi: http://dx.doi.org/10.1101/049288
upravit: v květnu 2016 byl David aktualizován!
https://david.ncifcrf.gov/content.jsp?file=release.html















