Frontiers in Endocrinology
Einleitung
Das Tumorsuppressor-Gen TP53 ist seit seiner Entdeckung vor fast 40 Jahren das am stärksten untersuchte menschliche Gen (1). Der Hauptgrund für diesen Status ist die entscheidende Rolle, die p53 bei der Verhinderung der Krebsentwicklung spielt, und es wird allgemein als „Wächter des Genoms“ angesehen.,“Seit einiger Zeit wird allgemein angenommen, dass p53 aufgrund seiner Fähigkeit, Apoptose, Zellzyklusstillstand und Seneszenz präkanzeröser Zellen zu induzieren, eine Rolle bei der Tumorsuppression spielt (2). Es wird jedoch zunehmend klar, dass p53 viele andere Wege in der Zelle reguliert und dass diese anderen Wege auch eine Rolle bei der Fähigkeit von p53 spielen, als Tumorsuppressor zu fungieren (3). Insbesondere die Rolle von p53 bei der Regulation von Genen, die am Stoffwechsel und an der Ferroptose beteiligt sind, war an seiner Fähigkeit beteiligt, die Tumorentwicklung zu unterdrücken., Ferroptose ist ein neuartiger Zelltodweg, der erstmals 2012 charakterisiert wurde und am besten als eisenabhängige, caspaseunabhängige Form des Zelltods beschrieben werden kann, die durch die Bildung von Lipidperoxidation angetrieben wird (4). Insbesondere zwei Mausmodelle, die engineered Mutationen in p53 enthalten, die die Fähigkeit von p53, Apoptose und Seneszenz zu induzieren, eliminieren, behalten beide die Fähigkeit, die spontane Tumorentwicklung zu unterdrücken; Beide Mutanten behalten die Fähigkeit, Gene im Stoffwechsel und in der Ferroptose zu transaktivieren (5, 6)., Eine Zusammenfassung der Daten, die p53 in die Regulation des Stoffwechsels und der Ferroptose einbeziehen, ist nachstehend aufgeführt.
Wildtyp (WT) p53 reguliert positiv die oxidative Phosphorylierung und unterdrückt den Glukosestoffwechsel
Wildtyp p53 reguliert die metabolische Vielseitigkeit von Zellen, indem es die mitochondriale Atmung gegenüber der Glykolyse begünstigt, zum Teil über die Transaktivierung von SCO2 (Cytochrom-c-Oxidase-Assembly), das eine direkte Rolle bei der oxidativen Phosphorylierung spielt (7)., p53 reguliert auch direkt die Transaktivierung von GLS2 (Glutaminase 2); Dieses Enzym ermöglicht den Glutaminverbrauch als Energiequelle für die Mitochondrien (8). Darüber hinaus reguliert WT p53 die Glykolyse negativ, indem es die Glukosetransporter GLUT1 und GLUT4 transkriptionell unterdrückt und RRAD und TIGAR transaktioniert; beide sind Inhibitoren der Glykolyse (9-11). Schließlich bindet und hemmt p53 auch direkt das Enzym Glucose-6-Phosphat-Dehydrogenase und unterdrückt so den Glukosestoffwechsel (12)., Aus diesen und anderen Studien geht hervor, dass p53 in normalen, unbelasteten Organismen den Stoffwechselzustand in einer Zelle direkt reguliert (Abbildung 1). Es überrascht nicht, dass dieses Gen und viele seiner Regulatoren an Stoffwechselerkrankungen, einschließlich Fettleibigkeit und Diabetes, beteiligt sind (13).
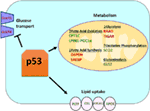
Abbildung 1. Die Rolle von Wildtyp (WT) p53 im Stoffwechsel. Gene, die durch p53 positiv reguliert werden, sind grün dargestellt, und Gene, die durch p53 negativ reguliert werden, sind rot dargestellt., p53 hemmt Glukosetransport, Glykolyse und Fettsäuresynthese, während es Lipidaufnahme, Fettsäureoxidation, oxidative Phosphorylierung und Glutaminolyse fördert.
Mutant p53 Reguliert positiv den Warburg-Stoffwechsel (aerobe Glykolyse)
Im Gegensatz zur Funktion von WT p53 begünstigt Mutant p53 in Tumorzellen die aerobe Glykolyse, zum Teil durch Verbesserung des Handels des Glukosetransporters GLUT1 mit der Plasmamembran, wodurch der Glukosimport erhöht wird (14, 15)., Nach der Mutation von p53 begünstigen die reduzierten SCO2-und GLS2-Spiegel und die erhöhten GLUT1-und GLUT4-Spiegel die aerobe Glykolyse gegenüber der oxidativen Phosphorylierung. Auf diese Weise wird angenommen, dass mutiertes p53 zur Neigung von Tumorzellen beiträgt, aerobe Glykolyse zugunsten der oxidativen Phosphorylierung oder des sogenannten Warburg-Metabolismus zu nutzen (15). Eines der Kennzeichen von Krebs ist der deregulierter Stoffwechsel, der sich im Allgemeinen durch diesen Wechsel von aerober Glykolyse zur oxidativen Phosphorylierung zeigt., Obwohl dies zu einer geringeren und weniger effizienten ATP-Ausbeute führt, wird angenommen, dass Krebszellen davon profitieren, indem glykolytische Zwischenprodukte auf biosynthetische Wege umgeleitet werden, die für eine schnelle Zellteilung erforderlich sind (16). Dieser metabolische Schalter führt auch zu einer verminderten Mitochondrien-vermittelten Apoptose und einer effizienteren Signalisierung durch verfügbare Metaboliten in Krebszellen (17).
Eine häufige genetische Variante in TP53 beeinflusst ihre Funktion im Stoffwechsel
Es gibt einen gemeinsamen codierenden Regionenpolymorphismus von p53 bei Codon 72, der entweder für Prolin (P72) oder Arginin (R72) kodiert., Diese Aminosäurevariation kann die p53-Funktion in Bezug auf das Zellverhalten nach Stress beeinflussen. Als Reaktion auf DNA-Schäden löst die P72-Variante von p53 überwiegend einen Zellzyklusstillstand aus, während die R72-Variante überwiegend den Zelltod oder die Apoptose induziert (18, 19). Trotz dieser Funktionsunterschiede war die Codon 72-Variation nicht konsistent mit der Krebsanfälligkeit assoziiert (20). Im Gegensatz dazu ist dieser Polymorphismus in Studien am Menschen signifikant mit einem erhöhten Body-Mass-Index und einem erhöhten Risiko für Diabetes verbunden (21, 22)., Diese Prämisse wird durch Studien an Mäusen gestützt, bei denen ein Mausmodell für diese Codon 72-Varianten bei Mäusen mit der R72-Variante im Vergleich zu P72 einen erhöhten fettreichen diätinduzierten Diabetes zeigt. In diesen Studien wurden die p53-Zielgene TNFa und NPC1L1 als kritische Regulatoren für den Anstieg der ernährungsinduzierten Fettleibigkeit bei R72-Mäusen identifiziert (23). Interessanterweise hat sich auch gezeigt, dass die R72-Variante als Reaktion auf Nährstoffmangel ein erhöhtes Überleben der Zellen ermöglicht (24)., Diese Ergebnisse haben zu der Hypothese geführt, dass die R72-Variante von p53 entstand und ausgewählt wurde, da Populationen nach Norden wanderten, wo kaltes Wetter eine erhöhte Fettansammlung erfordern würde, aber wo das Überleben als Reaktion auf Nährstoffmangel ebenfalls unter Auswahl wäre (24).
p53 reguliert den Fettstoffwechsel
Obwohl p53 für die Regulierung der Glykolyse und des Zitronensäurekreislaufs bekannt ist, spielt p53 nachweislich auch eine Rolle bei der Regulierung des Fettstoffwechsels (25)., Es wird angenommen, dass WT p53 die Fettsäureoxidation verbessert und gleichzeitig die Fettsäuresynthese hemmt, wodurch es als negativer Regulator der Lipidsynthese wirkt (25). Es gibt mehrere p53-Zielgene mit Rollen im Fettstoffwechsel. Sanchez-Macedo und Kollegen zeigten, dass Carnitinpalmitoyltransferase 1C (CPT1C) transkriptional durch p53 reguliert wird; Dieses Enzym hilft beim Transport von aktivierten Fettsäuren zu den Mitochondrien., Zur Unterstützung einer Rolle dieses p53-regulierten Gens bei Krebs zeigte diese Gruppe, dass Cpt1c-defiziente Mäuse eine verzögerte Tumorentwicklung und höhere Überlebensraten aufweisen (26). Lipin 1 (LPIN1) ist ein weiteres p53-Zielgen; LPIN1 ist für eine ordnungsgemäße Adipozytenentwicklung notwendig und wird unter nährstoffarmen Bedingungen induziert (27). Finck und Kollegen zeigten, dass LPIN1 mit PGC-1α, einem anderen bekannten p53-Zielgen mit einer Rolle im Stoffwechsel, interagiert und dass diese Wechselwirkung die Expression von Genen aktiviert, die an der Förderung der Fettsäureoxidation beteiligt sind (28).,
Neben der direkten Regulierung der Transkription von Genen, die am Fettstoffwechsel beteiligt sind, kann p53 auch den Fettstoffwechsel auf eine Weise regulieren, die eine direkte Protein–Protein-Interaktion beinhaltet. Beispielsweise bindet die Glucose-6-Phosphat-Dehydrogenase, die das geschwindigkeitslimitierende Enzym im Pentosephosphatweg ist, an p53 und wird direkt gehemmt, was zu einer verminderten NADPH-Produktion und folglich zu einer verminderten Fettsäuresynthese führt (12)., Die Familie der Transkriptionsfaktoren der Sterol-regulatorischen elementbindenden Proteine (SREBP) moduliert die Expression von Genen, die an der Cholesterin -, Fettsäure -, Triacylglycerin-und Phospholipidsynthese beteiligt sind (29-31). WT p53 unterdrückt die SREBP-Funktion (32), während mutierte Formen von p53 direkt an SREBP binden und ihre Transkriptionsfunktion verbessern, was zu einer erhöhten SREBP-Aktivität bei menschlichen Tumoren führt (33, 34). Folglich ist mutiertes p53 mit einer höheren Expression von Sterol-Biosynthesegenen in menschlichen Brusttumoren korreliert (34, 35)., Schließlich ist AMP-aktivierte Proteinkinase (AMPK) ein Enzym, das unter niedrigem Nährstoffgehalt oder Energiestress aktiviert wird und bekanntermaßen die Fettsäuresynthese durch Wechselwirkung mit Acetyl-CoA-Carboxylase und SREBP-1 hemmt (36, 37). Zhou und Kollegen zeigten, dass mutiertes p53 bevorzugt an AMPK bindet und es hemmt, was zu einer erhöhten Fettsäuresynthese führt. Infolgedessen führen mutierte p53-Proteine zu einer erhöhten AMPK-Signalgebung und tragen zum invasiven Zellwachstum von Tumorzellen bei (33). Ein weniger erforschtes Gebiet ist die Rolle von p53 beim Lipidtransport., Es wurde gezeigt, dass p53 transkriptionell Apolipoprotein B (ApoB) und APOB2-Enzymkomplex 1 reguliert, was auf die Rolle von p53 bei der Regulierung atherogener Lipoproteine hinweist (38). Die Mikroarray-Analyse menschlicher Leber abgeleiteter Zellen identifizierte Phospholipid-Transferprotein, ATP-bindende Kassette A12 und Carboxylesterlipase als drei p53-Zielgene, die alle eine Rolle beim Lipidtransport spielen (39, 40)., Insgesamt ist zwar klar, dass p53 eine Schlüsselrolle bei der Vermittlung der Lipidsynthese und des Stoffwechsels spielt, der Beitrag dieses Weges und dieser p53-Zielgene zur Tumorsuppression durch p53 muss jedoch noch bestimmt werden (Abbildung 1).
Ferroptose ist ein neuartiger Zelltodweg, der durch Lipidperoxidation angetrieben wird
Im Jahr 2012 entdeckten Dixon und Kollegen eine neuartige Form des regulierten Zelltods namens Ferroptose. Ferroptose ist eine eisenabhängige, caspaseunabhängige Form des Zelltods, die aus der Akkumulation oxidierter Lipide resultiert (4, 41)., Dieser Prozess wird durch die Inaktivierung der Glutathionperoxidase 4 (GPX4) vorangetrieben, einem Enzym, das für die Umwandlung tödlicher Lipidhydroperoxide in nicht toxische Lipidalkohole verantwortlich ist, für deren Funktion Glutathion erforderlich ist (41). Es wird angenommen, dass die Peroxidation mehrfach ungesättigter Fettsäuren (PUFAs) der treibende Impuls für den Zelltod durch Ferroptose ist. PUFAs enthalten bis-Allylprotonen, die leicht abstrahiert werden können und Radikale produzieren, die mit Sauerstoff reagieren, mehr Radikale erzeugen und zu einer Kettenreaktion von lipidreaktiven Sauerstoffspezies führen (42)., Der genaue Mechanismus des Zelltodes durch Ferroptose bleibt unbekannt, aber eine Hypothese ist, dass der Lipidschaden zur Zerstörung der Plasmamembran führt (43). Es wurde spekuliert, dass Ferroptose ein Mechanismus der Tumorunterdrückung sein könnte, der Zellen eliminiert, denen Nährstoffe entzogen sind oder die einem Umweltstress oder einer Infektion ausgesetzt waren.,
Pharmakologische Regulation der Ferroptose
Ferroptose kann unter Verwendung von Inhibitoren des Systems xc− wie Erastin-oder Analoga wie Glutamat und Sorafenib induziert werden, die den Import von Cystin hemmen, was zu erschöpftem Glutathion und anschließender Inaktivierung von GPX4 führt. Alternativ kann ferroptosis durch (1S,3R)-RSL3 (im Folgenden als RSL3 bezeichnet) induziert werden, welches GPX4 direkt bindet und hemmt (4, 5, 42). Buthionsulfoximin, FIN56, FINO2, CCl4 und Cisplatin sind weitere Wirkstoffe, die nachweislich Ferroptose in Zellen induzieren., Der Tod durch Ferroptose kann durch Unterdrückung der Lipidperoxidation verhindert werden, was durch Verwendung lipophiler Antioxidantien wie Ferrostatin-1, Liproxstatin-1 oder Vitamin E erreicht werden kann E. Eisenchelatoren wie Deferoxamin oder Cicloprox sind ein weiteres Mittel zur Unterdrückung der Ferroptose durch Verringerung des Eisenspiegels. Der Abbau von PUFAs oder die Zugabe von einfach ungesättigten Fettsäuren zu Zellkulturmedien kann Zellen auch vor Ferroptose retten (42, 44).,
Ferroptose ist an der p53-vermittelten Tumorsuppression beteiligt
2012 entwickelten Gu und Kollegen ein Mausmodell, bei dem drei normalerweise acetylierte Lysin-Reste in der DNA-Bindungsdomäne von p53 zu Arginin mutiert waren und daher nicht acetyliert werden konnten; Diese Maus wird als 3KR-Maus bezeichnet. Insbesondere können Zellen der 3KR-Maus keine p53-abhängige Apoptose, Zellzyklusstillstand oder Seneszenz durchlaufen, und tatsächlich kann die 3KR-Mutante von p53 die Mehrheit der p53-Zielgene nicht transaktivieren., Interessanterweise entwickelt dieses Mausmodell nicht spontan Krebs, was bedeutet, dass p53 die Tumorentwicklung unabhängig von Seneszenz oder Apoptose unterdrücken könnte (45). Diese Gruppe fand heraus, dass das mutierte 3KR-Protein die Fähigkeit behält, sich einer Ferroptose zu unterziehen und den Cystin-Stoffwechsel zu regulieren, indem es die Expression des Cystin-Proteins SLC7A11 reguliert; Dies deutete darauf hin, dass die Ferroptose ein Weg sein könnte, der der p53-vermittelten Tumorunterdrückung zugrunde liegt., Wenn Wildtyp und 3KR MEFs mit dem Ferroptose-Induktor Erastin behandelt wurden, wurden fast 50% Zelltod beobachtet, während p53 und MEFs 20% Zelltod zeigten; Dies zeigt, dass p53 Zellen für Ferroptose sensibilisiert und dass auch andere Schlüsselregulatoren eine Rolle bei der Ferroptose spielen (5). Anschließend identifizierten Gu und Kollegen eine zusätzliche Acetylierungsstelle an Lysin 98 von p53, und sie erzeugten ein Mausmodell, in dem alle vier Acetylierungsstellen zu Arginin mutiert waren (4KR)., Interessanterweise war die 4KR-Mutante nicht in der Lage, Gene zu regulieren, die an Ferroptose wie SLC7A11 beteiligt sind, und im Gegensatz zur 3KR-Mutante war sie nicht in der Lage, die Tumorentwicklung zu unterdrücken (46). Obwohl derzeit korrelativ, implizieren diese Daten die Rolle von p53 bei der Ferroptose in seiner Fähigkeit, die Tumorentwicklung zu unterdrücken.
In nicht transformierten Zellen reguliert p53 die Ferroptose positiv
Zusätzlich zu SLC7A11 wurden mehrere andere direkte p53-Zielgene entdeckt, die eine Rolle bei der Ferroptose spielen. Dazu gehören GLS2, PTGS2 und SAT1., Studien aus zwei getrennten Gruppen unterstützen die Rolle von GLS2 bei der Ferroptose, von der bekannt ist, dass sie Glutathion senkt und den zellulären ROS-Spiegel erhöht. Jiang und Kollegen verwendeten Ferroptose-Inhibitoren in Kombination mit Glutaminolyse-Inhibitoren, um die Erastin-induzierte Ferroptose zu hemmen, wodurch gezeigt wurde, dass Ferroptose Glutaminolyse und GLS2 erfordert (47). Murphy und Kollegen zeigten, dass eine polymorphe Variante von p53 in der Lage war, Wachstumsstillstand und Seneszenz sowohl in menschlichen als auch in murinen Zellen zu induzieren, SLC7A11 jedoch nicht unterdrückten oder GLS2 transaktivierten., Diese Variante war bei der Induktion von Ferroptose und der Unterdrückung der Tumorentwicklung deutlich beeinträchtigt, was wiederum die Rolle von p53 bei der Ferroptose-vermittelten Tumorsuppression implizierte (48). Ein weiteres p53-Zielgen mit einer Rolle bei der Ferroptose ist PTGS2, ein Gen, das für das Enzym Cyclooxygenase-2 kodiert. Stockwell und Kollegen zeigten zunächst, dass die Induktion von Ferroptose unter Verwendung von Erastin und RSL3 zur Hochregulation von PTGS2 führte (41). Insbesondere wurde PTGS2 nicht durch Ferroptose-Induktoren in p53-Null-Zellen hochreguliert, was darauf hindeutet, dass diese Regulierung p53-abhängig ist (5)., Gegenwärtig wird die Upregulation von PTGS2 häufig als Ferroptosemarker verwendet (5, 41).
Eine aktuelle Studie der Gu-Gruppe zeigte, dass das p53-Zielgen SAT1 die Ferroptose reguliert (49). Die Autoren identifizierten SAT1 als direktes Ziel von p53 und zeigten, dass die Stummschaltung von SAT1 den Zelltod reduzierte, der durch reaktive Sauerstoffspezies in Zellen mit WT p53 induziert wurde, in p53-Null-Zellen jedoch keine Wirkung hatte. Mechanistisch zeigte diese Gruppe, dass SAT1 das Niveau und die Aktivität von Arachidonat-15-Lipoxygenase erhöht, einem eisenbindenden Enzym, das PUFAs oxidiert und die Lipidperoxidation erhöht., Insbesondere zeigte diese Studie, dass weder p53 noch SAT1 allein ausreichen, um eine Ferroptose auszulösen. Stattdessen stimmen die kombinierten Daten eher mit der Prämisse überein, dass p53 aufgrund der Regulierung von Genen, die zur Ferroptose beitragen, die Empfindlichkeit von Zellen gegenüber diesem Weg reguliert und nicht direkt Ferroptose induziert. Ob p53 andere an der Ferroptose beteiligte Gene reguliert, bleibt abzuwarten (Abbildung 2).
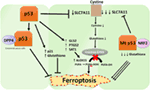
Abbildung 2. Die verschiedenen Rollen von p53 bei Ferroptose., Die Hemmung der Glutathionperoxidase 4 (GPX4), dem Schlüsselenzym, das die Umwandlung von mehrfach ungesättigten Fettsäuren (PUFAs), die Peroxide enthalten, in Alkohole katalysiert, ist der Haupttreiber der Ferroptose. Je nach Kontext kann p53 die Ferroptose (z. B. in Darmkrebszellen) unterdrücken oder die Ferroptose fördern. Mutiertes p53 sensibilisiert Zellen noch mehr für Ferroptose als Wildtyp p53.,
In einigen Zellen reguliert p53 die Ferroptose negativ
Eine kürzlich von Tarangelo und Kollegen veröffentlichte Studie zeigt, dass p53 die Ferroptose in Krebszellen negativ reguliert (50). Diese Gruppe fand heraus, dass die Vorbehandlung von Zellen mit Nutlin-3, einer Verbindung, die p53 stabilisiert, den Beginn der Ferroptose bei verschiedenen Zelltypen verzögert. Das verzögerte einsetzen der ferroptosis wurde gefunden hängen von CDKN1A (encoding p21), eine wichtige p53-transkriptionelle Ziel., Der Mechanismus, durch den p21 die Ferroptose verzögert, muss noch geklärt werden, aber es wird angenommen, dass die Erhaltung des intrazellulären Glutathions einen Beitrag zur verminderten Ferroptoseempfindlichkeit leisten kann. Die Autoren kommen zu dem Schluss, dass die p53-p21-Achse es Krebszellen ermöglicht, unter Stoffwechselstressbedingungen wie Cystin-Deprivation zu überleben, indem sie den Beginn der Ferroptose unterdrücken (50). Eine kürzlich durchgeführte Studie zeigte, dass p53 die Ferroptose in Darmkrebszellen hemmt, indem es an das Enzym Dipeptidylpeptidase-4 (DPP4) bindet, das ein Modulator der Ferroptose und des Fettstoffwechsels ist., Mechanistisch zeigte diese Studie, dass p53 die Ferroptose antagonisiert, indem DPP4 in einem nuklearen enzymatischen inaktiven Pool sequestriert wird. In Abwesenheit von p53 kann DPP4 frei mit einem Komplex ohne SAUERSTOFF interagieren und ihn bilden; Dies führt zu einer erhöhten Lipidperoxidation und Ferroptose. Die Hemmung von DPP4 unterdrückt die Ferroptose signifikant, während die Überexpression von DPP4 die Erastin-Empfindlichkeit auslöst, insbesondere in p53-erschöpften Zellen (51). Die bidirektionale Kontrolle der Ferroptose durch p53 durch transkriptionsabhängige und transkriptionsunabhängige Mechanismen kann kontext-oder zelltypabhängig sein (Abbildung 2).,
Der P47S-Polymorphismus von TP53 beeinflusst Ferroptose und Tumorunterdrückung
Zusätzlich zu Missense-Mutationen gibt es mehrere funktionell signifikante Single-Nucleotid-Polymorphismen (SNPs) im TP53-Gen und anderen Proteinen, von denen bekannt ist, dass sie diesen Weg regulieren (wie MDM2 und MDM4). Die Pro47Ser-Variante (im Folgenden S47) ist die zweithäufigste SNP, die in der p53-kodierenden Region (nach Pro72Arg) gefunden wird und die Aminosäuresequenz des Proteins verändert., Um die Auswirkungen dieser Variante auf die p53-Funktion und das Krebsrisiko besser aufzuklären, erzeugte die Murphy-Gruppe ein humanisiertes p53-Knock-in-Mausmodell, bei dem Exons 4-9 von murinem p53 durch humane p53-Exons ersetzt wurden, die entweder den Wildtyp oder die S47-Variante enthielten (52-55). Die Mehrheit der S47-Mäuse entwickelte spontan Tumore verschiedener histologischer Typen, insbesondere Leberkrebs, im Alter zwischen 12 und 18 Monaten, im Gegensatz zu WT p53-Mäusen (48)., In mausembryonalen Fibroblasten und humanen lymphoblastoiden Zelllinien zeigte die S47-Variante einen gestörten programmierten Zelltod als Reaktion auf Cisplatin und andere genotoxische Belastungen. Mechanistisch gesehen ist die S47-Variante für die Transaktivierung von am Stoffwechsel beteiligten Genen wie Gls2 (Glutaminase 2) und Sco2 (48) defekt. In Übereinstimmung mit der Rolle von Gls2 bei der Ferroptose stellte diese Gruppe fest, dass S47-Zellen gegenüber den Ferroptose-induzierenden Wirkstoffen Erastin und RSL3 deutlich resistent waren (47, 48). Dieser Defekt kann zu dem bei S47-Mäusen beobachteten tumoranfälligen Phänotyp beitragen.,
Mutant p53 sensibilisiert Tumorzellen für Ferroptose
Wildtyp p53 reguliert negativ die Expression des Cystin-Proteins SLC7A11, der die Empfindlichkeit gegenüber Ferroptose hemmt (5). Obwohl diese Regulation in normalen Zellen auftritt, scheinen in Tumorzellen andere Mediatoren von SLC7A11 bei der Regulation dieses Gens zu überwiegen. Zum Beispiel kann der Master-antioxidative Transkriptionsfaktor NRF2 auch die Expression von SLC7A11 auf Transkriptionsebene regulieren, und NRF2 wurde als Schlüsselakteur beim Schutz von Krebszellen vor Ferroptose in Verbindung gebracht., Beispielsweise erhöht die Hemmung von NRF2 in hepatozellulären Krebszellen die Antikrebsaktivität von Erastin und Sorafenib in vivo (56). Mutierte Formen von p53 können die NRF2-Funktion durch direkte Wechselwirkung hemmen, und eine Gruppe fand heraus, dass Tumore mit mutiertem p53 sehr niedrige SLC7A11-Spiegel enthalten und somit eine erhöhte Empfindlichkeit gegenüber Ferroptose zeigen. Insbesondere führte die Überexpression von SLC7A11 in mutierten p53-Modellen zu Arzneimittelresistenz, was darauf hindeutet, dass die Expression von SLC7A11 berücksichtigt werden muss, wenn mutierte p53-gesteuerte Krebsarten mit Ferroptose-induzierenden Verbindungen angestrebt werden (57)., Zur Unterstützung dieser Prämisse zeigten jüngste Arbeiten an kolorektalem (CRC) Krebs, bei denen Mutation oder Deletion von p53 ein häufiges Ereignis ist, dass humane CRC-Zelllinien, die mutierte p53 enthalten, im Vergleich zu CRC-Zellen mit Erastin-vermitteltem Zelltod weitaus empfindlicher auf den Zelltod reagierten WT p53. Um diese Ergebnisse zu validieren, zeigten sie, dass das Anklopfen einer p53-Hotspot-Mutation in HCT116-und SW48-Zellen die Empfindlichkeit gegenüber Erastin wiederherstellte (51). Diese Daten zeigen einen neuartigen Mechanismus auf, mit dem Krebsarten, die von mutiertem p53 angetrieben werden, mithilfe einer gezielten Therapie ausgenutzt werden können.,
Schlussfolgerung
Die Rolle von p53 im Stoffwechsel ist ziemlich klar und möglicherweise sogar intuitiv offensichtlich: WT p53 begrenzt den Glukosestoffwechsel und die Lipidsynthese, während mutiertes p53 das Gegenteil zu tun scheint. Der Beitrag seiner metabolischen Rolle zur Tumorsuppression durch p53 und zur Fähigkeit von mutiertem p53, die Tumorprogression voranzutreiben, muss eindeutig nachgewiesen werden. Die Rolle von p53 bei der Regulation der Ferroptose und der Beitrag dieser Funktion zur Tumorsuppression ist noch weniger klar., Während überzeugende Daten aus Mausmodellen die Prämisse stützen, dass p53 die Empfindlichkeit von Zellen gegenüber Ferroptose reguliert, kann dies auf die Fähigkeit von basalem p53 beschränkt sein, die spontane Tumorentwicklung zu unterdrücken, und in Onkogen-gestressten Mausmodellen ist es klar, dass Seneszenz und Apoptose die vorherrschende Rolle spielen. In ähnlicher Weise kann p53 die Ferroptosesensitivität zelltypspezifisch regulieren. Weitere Studien an Tiermodellen mit Aufmerksamkeit auf Ferroptose in verschiedenen Geweben müssen durchgeführt werden, um die Rolle von p53 bei Ferroptose und Ferroptose bei der Tumorsuppression besser zu verstehen., Darüber hinaus muss eine klarere Vorstellung davon, welche p53-Zielgene eine Rolle bei der Empfindlichkeit gegenüber Ferroptose spielen, erreicht werden. Die Lösung dieser Fragen sollte dringend benötigte neue Wege zur Bekämpfung von Tumoren mit mutiertem p53 bieten.
Autor Beiträge
KG, SB, TB, AB-K, C-PK, und MM, schrieb ein bis zwei Absätze dieses Artikels. KG und SB haben die Figur gemacht. KG und MM umrissen das Kapitel.,
Interessenkonflikterklärung
Die Autoren erklären, dass die Untersuchung ohne kommerzielle oder finanzielle Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Der Reviewer OAF und der Editor erklärten ihre gemeinsame Zugehörigkeit.
Anerkennungen
Die in dieser Veröffentlichung berichteten Forschungen wurden von den National Institutes of Health unter den Zulassungsnummern CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) und T32 CA009171 (TB) unterstützt., Der Inhalt liegt allein in der Verantwortung der Autoren und stellt nicht unbedingt die offiziellen Ansichten der National Institutes of Health dar.
3. Humpton TJ, Vousden KH. Regulierung des Zellstoffwechsels und der Hypoxie durch p53. Kalter Frühling im Harz (2016) 6(7):211-30. doi: 10.1021 / cshperspect.a026146
CrossRef Volltext | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., Tumorsuppressor p53 reguliert negativ die durch Hypoxie stimulierte Glykolyse durch seine Ziel-RRAD. Oncotarget (2014) 5(14):5535-46. doi:10.18632/oncotarget.2137
PubMed Abstract | CrossRef Volltext | Google Scholar
17. Lee M, Yoon JH. Metabolisches Wechselspiel zwischen Glykolyse und mitochondrialer Oxidation: der umgekehrte Warburg-Effekt und seine therapeutische Implikation. World J Biol Chem (2015) 6(3):148-61. doi:10.4331/wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., Die Erschöpfung des neuartigen p53-Zielgens Carnitinpalmitoyltransferase 1C verzögert das Tumorwachstum im Neurofibromatose-Typ-I-Tumormodell. Zelltod unterschiedlich (2013) 20(4):659-68. doi:10.1038/cdd.2012.168
PubMed Abstract | CrossRef Volltext | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation von Sterol-Antwortelement-bindenden Proteinen und nachgeschalteten Effektoren bei Prostatakrebs während des Fortschreitens zur Androgenunabhängigkeit., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in-Mäuse mit einem chimären humanen/murinen p53-Gen entwickeln sich normal und zeigen Wildtyp-p53-Reaktionen auf DNA-schädigende Mittel: ein neues biomedizinisches Forschungswerkzeug. Onkogen (2001) 20(3):320-8. doi:10.1038/sj.onc.1204080
PubMed Abstract | CrossRef Volltext | Google Scholar
57. Liu DS, Duong CP, Haupt S, Montgomery, KG, Haus CM, Azar WJ, et al. Die Hemmung der xC-/Glutathion-Achse des Systems zielt selektiv auf Krebserkrankungen mit mutierter p53-Akkumulation ab., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















