– Genom-Spot
Pathway-Analysen geworden wie ein gemeinsames Vorgehen in der Bioinformatik, insbesondere in der Untersuchung der Genexpression. Wenn Sie sich eine Umfrage der letzten Papiere ansehen, scheint es eine Reihe von Möglichkeiten zu geben, dies zu tun. In diesem Beitrag werde ich die Unterschiede zwischen zwei häufig verwendeten Tools diskutieren; DAVID und GSEA.

Die Konzepte, die hinter den beiden algorithmen sind sehr unterschiedlich., DAVID ermittelt Überlappungen zwischen benutzerdefinierten Genlisten und den kuratierten Datenbanken und sucht nach Überlappungen, die größer sind als zufällig erwartet. Sie können die Genauigkeit des Algorithmus verbessern, indem Sie eine Hintergrunddatei bereitstellen, die alle Gene enthält, die im Experiment berücksichtigt/erkannt wurden. Die vom Benutzer bereitgestellten Gensätze werden normalerweise durch Auswahl von Genen erzeugt, die eine Signifikanzschwelle überschreiten. Das DAVID-Verfahren ähnelt anderen verfügbaren wie Ingenuity, AmiGO und GeneGO., Die Auswahl der Signifikanzwerte ist weitgehend willkürlich, es ist jedoch üblich, den Schwellenwert auf FDR > p<0.05 zu setzen. Durch die Änderung des Schwellenwerts werden die Anreicherungsergebnisse erheblich verändert. Die verwendeten statistischen Tests umfassen hypergeometrisch, Fisher ‚ s exact und Chi-Squared.
Im Gegensatz zu den oben genannten Methoden, die Listen von Genen erfordern, die beliebige Schwellenwerte passieren, ist GSEA ein Werkzeug, das jeden Datenpunkt in seinem statistischen Algorithmus verwendet. In der“ klassischen “ Methode werden Gene von den meisten up-reguliert bis zu den meisten Down-reguliert eingestuft., Die Rangmetrik selbst variiert, aber zwei gültige Methoden verwenden den vorzeichenbehafteten p-Wert oder das niedrigere 90% – Konfidenzintervall der Falzänderung. Grundlage des Tests ist die Beurteilung, ob Mitglieder eines Gensatzes an einem Ende des Profils angereichert erscheinen. Um die Anreicherung zu testen, führt GSEA Permutationen des Profils durch und berechnet die Anreicherung des Gensatzes tausendmal oder öfter, um p-Werte empirisch abzuschätzen. Andere Tools, die das gesamte Profil verwenden, führen analytische Tests durch, wie den Wilcoxon-und Kolmogorov-Smirnov-Test.,
Jetzt, da wir die Hauptunterschiede in den Algorithmen verstehen, können wir beginnen, die Ergebnisse dieser beiden Techniken zu vergleichen. Ich habe einen Datensatz überarbeitet, den ich zuvor besprochen habe, Azacitidin-Behandlung in AML3-Zellen, die durch RNA-seq profiliert wurden. Ich werde die von DESeq generierten differentiellen RNA-seq-Expressionsdaten als Startpunkt verwenden, um DAVID und GSEA zu vergleichen.
Für DAVID habe ich eine Hintergrunddatei erstellt, die aus allen 19.030 Genen besteht, die im Experiment oberhalb der Nachweisschwelle nachgewiesen wurden. Die signifikant (FDR<0.,05) genlisten in der up (2502 Gene) und down (2907 Gene) Richtung wurden separat an DAVID 6.7 zur Analyse vorgelegt, wobei der Schwerpunkt nur auf KEGG-Pfaden lag.
Für GSEA habe ich das gleiche zuvor beschriebene Rangmetrikverfahren verwendet. Ich habe den klassischen GSEA-Algorithmus mit dem gsea2-2.2.2 verwendet.jar ausführbare Datei mit KEGG-Gensätzen von MSigDB v5.1.
In der folgenden Grafik habe ich die Überlappung statistisch signifikanter Gensätze (FDR<0.05) separat für regulierte Wege nach oben und unten bestimmt., Ich habe auch einen Schnittpunkt der Ergebnisse eingefügt, wenn kein Hintergrund angegeben ist (nur aus Neugier, kein empfohlenes Verfahren).
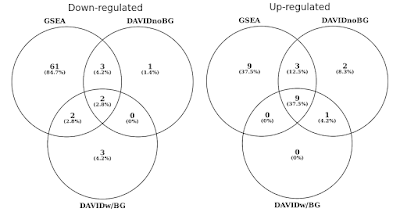
DAVID vs GSEA mit demselben Genexpressionsprofil. Zahlen stellen Gensätze dar, die signifikant angereichert sind (FDR<0.05).
Sie werden feststellen, dass GSEA sowohl in Aufwärts-als auch in Abwärtsrichtung im Vergleich zu DAVID signifikantere Ergebnisse identifiziert. Ich fand, dass dies auch in anderen Gensatzbibliotheken wie REACTOME und GO der Fall war., Der Unterschied in der Empfindlichkeit zwischen den beiden Werkzeugen läuft auf die verschiedenen Algorithmen hinaus, wobei DAVID eine diskrete Liste von Genen verwendet, im Vergleich zu GSEA, das jeden Datenpunkt verwendet. GSEA wird bei der Identifizierung von Gensätzen mit vielen Mitgliedern, die subtile Veränderungen in der Expression erfahren, viel empfindlicher sein. Einzeln sind diese Veränderungen statistisch nicht signifikant, aber wenn ein großer Teil der Gene in einer Gruppe die Expression ändert, könnte dieser Trend statistisch gesehen sehr signifikant sein.,
Zusätzlich zu dem empfindlicheren Algorithmus sind Davids Gensätze und Genanmerkungen wirklich veraltet. Die letzte Aktualisierung von DAVIDS vielen Datenquellen erfolgte Mitte bis Ende 2009. Daher werden viele Genzugriffe von Ensembl möglicherweise nicht unterstützt. In der Tat wurden aus meiner Hintergrunddatei, die 19.030 enthielt, nur 13.488 von DAVID erkannt. Das Fehlen aktualisierter Gensätze ist ebenfalls ein Problem, ebenso wie die ständig wachsende Anzahl von Zitaten, die DAVID immer noch erhält.,
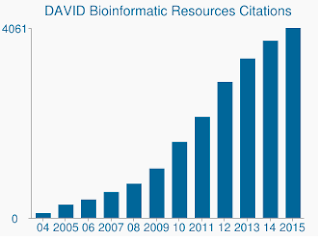
DAVID erhält immer noch Tausende von Zitaten pro Jahr, obwohl die zugrunde liegenden Daten veraltet sind.
Abschließend würde ich Forschern empfehlen, Tools zu verwenden, die ein ganzes Profil betrachten, um die Anreicherung zu bestimmen (GSEA, CAMERA, WilcoxGST, Pathifier), anstatt Gensätze für die Überlappungsanalyse einzureichen. Die Verwendung von DAVID könnte die schnelle und einfache Option sein, aber dies geht auf Kosten einer massiven Verringerung der Empfindlichkeit.
Weiter Lesen
JA Timmons, KJ Szkop, IJ Gallagher., Mehrere Quellen von Bias verwechseln funktionale Bereicherung Analyse von global-omics Daten. Genome biology, 2015. 16:186
Lina Wadi, Mona, Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Auswirkungen der Wissensakkumulation auf die Wegbereicherungsanalyse. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: Im Mai 2016 war DAVID aktualisiert!
https://david.ncifcrf.gov/content.jsp?file=release.html















