Frontiers in Endocrinology (Français)
Introduction
le gène suppresseur tumoral TP53 est le gène humain le plus étudié depuis sa découverte il y a près de 40 ans (1). La principale raison de ce statut est le rôle critique que joue p53 dans la prévention du développement du cancer, et il est largement considéré comme le « Gardien du génome.,” Depuis un certain temps, on croit généralement que le rôle de p53 dans la suppression tumorale est dû à sa capacité à induire l’apoptose, l’arrêt du cycle cellulaire et la sénescence des cellules précancéreuses (2). Cependant, il est maintenant de plus en plus clair que p53 régule de nombreuses autres voies dans la cellule et que ces autres voies jouent également un rôle dans la capacité de p53 à fonctionner comme suppresseur de tumeur (3). En particulier, le rôle de p53 dans la régulation des gènes impliqués dans le métabolisme et la ferroptose a été impliqué dans sa capacité à supprimer le développement tumoral., La ferroptose est une nouvelle voie de mort cellulaire caractérisée pour la première fois en 2012 et peut être mieux décrite comme une forme de mort cellulaire dépendante du fer et indépendante de la caspase entraînée par la formation de peroxydation lipidique (4). Plus précisément, deux modèles murins contenant des mutations modifiées dans p53 qui éliminent la capacité de p53 à induire l’apoptose et la sénescence conservent la capacité de supprimer le développement tumoral spontané; ces deux mutants conservent la capacité de transactiver les gènes dans le métabolisme et la ferroptose (5, 6)., Un résumé des données impliquant p53 dans la régulation du métabolisme et de la ferroptose est détaillé ci-dessous.
Wild-Type (WT) p53 régule positivement la Phosphorylation oxydative et supprime le métabolisme du Glucose
Wild-type p53 régule la polyvalence métabolique des cellules en favorisant la respiration mitochondriale par rapport à la glycolyse, en partie via la transactivation de la SCO2 (cytochrome C oxydase assembly), qui joue un rôle direct dans la phosphorylation oxydative (7)., p53 régule également directement la transactivation de la GLS2( Glutaminase 2); cette enzyme permet l’utilisation de la glutamine comme source d’énergie pour les mitochondries (8). En outre, WT p53 régule négativement la glycolyse en réprimant transcriptionnellement les transporteurs de glucose GLUT1 et GLUT4, et en transactivant RRAD et TIGAR; les deux sont des inhibiteurs de la glycolyse (9-11). Enfin, p53 se lie et inhibe directement l’enzyme glucose-6-phosphate déshydrogénase, supprimant ainsi le métabolisme du glucose (12)., Il ressort clairement de ces études et d’autres que dans les organismes normaux et non stressés, p53 régule directement l’état métabolique dans une cellule (Figure 1). Sans surprise, ce gène et plusieurs de ses régulateurs sont impliqués dans des maladies métaboliques, y compris l’obésité et le diabète (13).
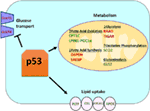
la Figure 1. Le rôle de type sauvage (WT) de la p53 dans le métabolisme. Les gènes régulés positivement par p53 sont indiquées en vert, et les gènes négativement régulée par p53 sont indiqués en rouge., p53 inhibe le transport du glucose, la glycolyse et la synthèse des acides gras alors qu’il favorise l’absorption des lipides, l’oxydation des acides gras, la phosphorylation oxydative et la glutaminolyse.
le Mutant p53 régule positivement le métabolisme de Warburg (glycolyse aérobie)
contrairement à la fonction de WT p53, le mutant p53 dans les cellules tumorales favorise la glycolyse aérobie, en partie en améliorant le trafic du transporteur de glucose GLUT1 vers la membrane plasmique, augmentant ainsi l’importation de glucose (14, 15)., Suite à la mutation de p53, les niveaux réduits de SCO2 et GLS2 et les niveaux accrus de GLUT1 et GLUT4 favorisent la glycolyse aérobie par rapport à la phosphorylation oxydative. De cette manière, on pense que le mutant p53 contribue à la propension des cellules tumorales à utiliser la glycolyse aérobie en faveur de la phosphorylation oxydative, ou métabolisme dit de Warburg (15). L’une des caractéristiques du cancer est le métabolisme dérégulé, généralement démontré par ce passage de la glycolyse aérobie à la phosphorylation oxydative., Bien qu’il en résulte un rendement en ATP plus faible et moins efficace, on pense que les cellules cancéreuses bénéficient en détournant les intermédiaires glycolytiques vers les voies biosynthétiques nécessaires à la division cellulaire rapide (16). Ce commutateur métabolique entraîne également une diminution de l’apoptose médiée par les mitochondries et une signalisation plus efficace par les métabolites disponibles dans les cellules cancéreuses (17).
une variante génétique commune dans TP53 influence sa fonction dans le métabolisme
Il y a un polymorphisme commun de région codante de p53 au codon 72, codant pour la proline (P72) ou l’arginine (R72)., Cette variation d’acide aminé peut avoir un impact sur la fonction p53 en ce qui concerne le devenir cellulaire après un stress. En réponse aux dommages à l’ADN, la variante P72 de p53 déclenche principalement l’arrêt du cycle cellulaire, tandis que la variante R72 induit principalement la mort cellulaire, ou l’apoptose (18, 19). Malgré ces différences de fonction, la variation du codon 72 n’a pas été systématiquement associée à la sensibilité au cancer (20). En revanche, dans les études humaines, ce polymorphisme est significativement associé à une augmentation de l’indice de masse corporelle et du risque de diabète (21, 22)., Cette prémisse est corroborée par des études chez la souris, où un modèle murin pour ces variantes du codon 72 montre une augmentation du diabète induit par l’alimentation riche en graisses chez les souris avec la variante R72, par rapport à P72. Dans ces études, Les gènes cibles P53 TNFa et NPC1L1 ont été identifiés comme régulateurs critiques de l’augmentation de l’obésité induite par l’alimentation chez les souris R72 (23). Fait intéressant, il a également été démontré que la variante R72 confère une survie accrue des cellules en réponse à la privation de nutriments (24)., Ces résultats ont conduit à l’hypothèse que la variante R72 de p53 est apparue et a été sélectionnée pour que les populations migrent vers le nord, où le temps froid nécessiterait une accumulation accrue de graisse, mais où la survie en réponse à la privation de nutriments serait également sous sélection (24).
P53 régule le métabolisme lipidique
bien que p53 soit bien connu pour réguler la glycolyse et le cycle de l’acide citrique, il a également été démontré que p53 joue un rôle dans la régulation du métabolisme lipidique (25)., On pense que WT p53 améliore l’oxydation des acides gras tout en inhibant la synthèse des acides gras, agissant ainsi comme un régulateur négatif de la synthèse lipidique (25). Il existe plusieurs gènes cibles p53 ayant des rôles dans le métabolisme des lipides. Sanchez-Macedo et ses collègues ont démontré que la carnitine palmitoyltransférase 1C (CPT1C) est régulée transcriptionnellement par p53; cette enzyme aide au transport des acides gras activés vers les mitochondries., À l’appui du rôle de ce gène régulé par p53 dans le cancer, ce groupe a montré que les souris déficientes en Cpt1c présentaient un développement tumoral retardé et des taux de survie plus élevés (26). La lipine 1 (LPIN1) est un autre gène cible p53; la LPIN1 est nécessaire au bon développement des adipocytes et est induite dans des conditions de faible teneur en nutriments (27). Finck et ses collègues ont montré que LPIN1 interagit avec PGC-1α, un autre gène cible p53 connu ayant un rôle dans le métabolisme, et que cette interaction active l’expression des gènes impliqués dans la promotion de l’oxydation des acides gras (28).,
en plus de réguler directement la transcription des gènes impliqués dans le métabolisme lipidique, p53 peut également réguler le métabolisme lipidique d’une manière impliquant une interaction protéine–protéine directe. Par exemple, la glucose-6-phosphate déshydrogénase, qui est l’enzyme limitant le débit dans la voie du pentose phosphate, se lie au p53 et est directement inhibée par celui-ci, ce qui entraîne une diminution de la production de NADPH et, par conséquent, une diminution de la synthèse des acides gras (12)., La famille des facteurs de transcription srebp (sterol regulatory element-binding proteins) module l’expression des gènes impliqués dans la synthèse du cholestérol, des acides gras, du triacylglycérol et des phospholipides (29-31). WT p53 réprime la fonction SREBP (32), tandis que les formes mutantes de p53 se lient directement à SREBP et améliorent leur fonction transcriptionnelle, conduisant à une augmentation de l’activité srebp dans les tumeurs humaines (33, 34). Par conséquent, le mutant p53 est corrélé à une expression plus élevée des gènes de biosynthèse des stérols dans les tumeurs mammaires humaines (34, 35)., Enfin, la protéine kinase activée par L’AMP (AMPK) est une enzyme qui est activée sous de faibles niveaux de nutriments ou un stress énergétique et qui est connue pour inhiber la synthèse des acides gras en interagissant avec l’acétyl-CoA-carboxylase et la SREBP-1 (36, 37). Zhou et ses collègues ont démontré que le mutant p53 se lie préférentiellement à L’AMPK et l’inhibe, ce qui entraîne une augmentation de la synthèse des acides gras. En conséquence, les protéines mutantes p53 conduisent à une augmentation de la signalisation AMPK, contribuant à la croissance cellulaire invasive des cellules tumorales (33). Un domaine moins exploré est le rôle du p53 dans le transport des lipides., Il a été démontré que la p53 régule transcriptionnellement l’apolipoprotéine B (apoB) et le complexe enzymatique d’édition apoB 1, ce qui indique le rôle de la p53 dans la régulation des lipoprotéines athérogènes (38). L’analyse par microréseaux de cellules dérivées du foie humain a identifié la protéine de transfert des phospholipides, la cassette de liaison à L’ATP A12 et la lipase de l’ester carboxylique comme trois gènes cibles p53 qui jouent tous un rôle dans le transport des lipides (39, 40)., Dans l’ensemble, bien qu’il soit clair que p53 joue un rôle clé dans la médiation de la synthèse lipidique et du métabolisme, la contribution de cette voie et de ces gènes cibles p53 à la suppression tumorale par p53 reste à déterminer (Figure 1).
la Ferroptose est une nouvelle voie de mort cellulaire entraînée par la peroxydation lipidique
en 2012, Dixon et ses collègues ont découvert une nouvelle forme de mort cellulaire régulée appelée ferroptose. La ferroptose est une forme de mort cellulaire dépendante du fer et indépendante de la caspase résultant de l’accumulation de lipides oxydés (4, 41)., Ce processus est entraîné par l’inactivation de la glutathion peroxydase 4 (GPX4), une enzyme responsable de la conversion des hydroperoxydes lipidiques mortels en alcools lipidiques non toxiques, ce qui nécessite du glutathion pour fonctionner (41). On pense que la peroxydation des acides gras polyinsaturés (AGPI) est le moteur de la mort cellulaire par ferroptose. Les AGPI contiennent des protons bis-allyliques qui peuvent facilement être abstraits et produisent des radicaux qui réagiront avec l’oxygène, créant plus de radicaux et entraînant une réaction en chaîne des espèces réactives de l’oxygène lipidique (42)., Le mécanisme exact de la mort cellulaire par ferroptose reste inconnu, mais une hypothèse est que la lésion lipidique entraîne la destruction de la membrane plasmique (43). Il a été spéculé que la ferroptose pourrait être un mécanisme de suppression tumorale qui fonctionne en éliminant les cellules qui sont privées de nutriments ou qui ont été exposées à un stress environnemental ou à une infection.,
régulation pharmacologique de la Ferroptose
La Ferroptose peut être induite à l’aide d’inhibiteurs de system xc− tels que l’érastine, ou d’analogues tels que le glutamate et le sorafénib, qui inhibent l’importation de la cystine, entraînant une diminution du glutathion et une inactivation ultérieure de GPX4. Alternativement, la ferroptose peut être induite par (1s, 3R)-RSL3 (ci-après dénommé RSL3), qui se lie directement à GPX4 (4, 5, 42) et l’inhibe. Buthione sulfoximine, FIN56, FINO2, CCl4 et cisplatine sont d’autres agents qui ont été démontrés pour induire la ferroptose dans les cellules., La mort par ferroptose peut être évitée en supprimant la peroxydation lipidique, ce qui peut être accompli en utilisant des antioxydants lipophiles, tels que la ferrostatine-1, la liproxstatine-1 ou la vitamine E. Les chélateurs de fer tels que la déféroxamine ou le cicloprox sont un autre outil utilisé pour supprimer la ferroptose en réduisant les niveaux de fer. L’appauvrissement des AGPI ou l’ajout d’acides gras monoinsaturés aux milieux de culture cellulaire peuvent également sauver les cellules de la ferroptose (42, 44).,
la Ferroptose est impliquée dans la Suppression tumorale médiée par la p53
en 2012, Gu et ses collègues ont développé un modèle murin dans lequel trois résidus de lysine normalement acétylés dans le domaine de liaison à L’ADN de la p53 ont été mutés en arginine et ne pouvaient donc pas être acétylés; cette souris est Notamment, les cellules de la souris 3kr sont incapables de subir une apoptose dépendante de p53, un arrêt du cycle cellulaire ou une sénescence, et en effet le mutant 3kr de p53 ne parvient pas à transactiver la majorité des gènes cibles de p53., Fait intéressant, Ce modèle murin ne développe pas spontanément de cancer, ce qui implique que p53 pourrait supprimer le développement tumoral indépendamment de la sénescence ou de l’apoptose (45). Ce groupe a constaté que la protéine mutante 3kr conserve la capacité de subir la ferroptose et de réguler le métabolisme de la cystine en régulant l’expression de L’importateur de cystine SLC7A11; ceci a suggéré que la ferroptose pourrait être une voie qui sous-tend la suppression tumorale médiée par p53., Lorsque les MEF de type sauvage et 3KR ont été traités avec L’inducteur de ferroptose Erastin, on a observé une mort cellulaire de près de 50% alors que les MEF nuls p53 présentaient une mort cellulaire de 20%; cela indique que p53 sensibilise les cellules à la ferroptose et que d’autres régulateurs clés jouent également un rôle dans la ferroptose (5). Par la suite, Gu et ses collègues ont identifié un site d’acétylation supplémentaire à la lysine 98 de p53, et ils ont généré un modèle murin dans lequel les quatre sites d’acétylation ont été mutés en arginine (4KR)., Fait intéressant, le mutant 4KR était incapable de réguler les gènes impliqués dans la ferroptose comme SLC7A11, et contrairement au mutant 3KR était incapable de supprimer le développement tumoral (46). Bien qu’actuellement corrélatives, ces données impliquent le rôle de p53 dans la ferroptose dans sa capacité à supprimer le développement tumoral.
dans les cellules non transformées, p53 régule positivement la Ferroptose
en plus de SLC7A11, plusieurs autres gènes cibles directs p53 ont été découverts pour jouer un rôle dans la ferroptose. Ceux-ci incluent GLS2, PTGS2, et SAT1., Des études de deux groupes distincts soutiennent le rôle de GLS2 dans la ferroptose, qui est connue pour diminuer le glutathion et augmenter les niveaux de ROS cellulaires. Jiang et ses collègues ont utilisé des inhibiteurs de la ferroptose combinés à des inhibiteurs de la glutaminolyse pour inhiber la ferroptose induite par L’Érastine, démontrant ainsi que la ferroptose nécessite la glutaminolyse et GLS2 (47). Murphy et ses collègues ont montré qu’une variante polymorphe de p53 était capable d’induire l’arrêt de la croissance et la sénescence dans les cellules humaines et murines, mais n’a pas réussi à réprimer SLC7A11 ou à transactiver GLS2., Cette variante a été fortement altérée pour induire la ferroptose et supprimer le développement tumoral, impliquant ainsi à nouveau le rôle de p53 dans la suppression tumorale médiée par la ferroptose (48). Un autre gène cible p53 ayant un rôle dans la ferroptose est PTGS2, un gène codant pour l’enzyme cyclooxygénase-2. Stockwell et ses collègues ont d’abord montré que L’induction de la ferroptose à L’aide D’Erastin et de RSL3 entraînait la régulation à la hausse de PTGS2 (41). Notamment, PTGS2 n’a pas été régulé à la hausse par les inducteurs de ferroptose dans les cellules p53-nulles, ce qui suggère que cette régulation dépend de p53 (5)., Actuellement, la régulation à la hausse de PTGS2 est largement utilisée comme marqueur de ferroptose (5, 41).
Une étude récente du groupe Gu a montré que le gène cible P53 SAT1 régule la ferroptose (49). Les auteurs ont identifié SAT1 comme une cible directe de p53 et ont montré que le silençage de SAT1 réduisait la mort cellulaire induite par les espèces réactives de l’oxygène dans les cellules avec P53 en poids, mais n’avait aucun effet dans les cellules p53-nulles. Mécaniquement, ce groupe a montré que SAT1 augmente le niveau et l’activité de l’arachidonate 15-lipoxygénase, une enzyme liant le fer qui oxyde les AGPI et augmente la peroxydation lipidique., Notamment, cette étude a montré que ni p53 ni SAT1 seuls ne semblent être suffisants pour induire la ferroptose. Au lieu de cela, les données combinées sont plus cohérentes avec la prémisse que p53, en vertu de la régulation des gènes qui contribuent à la ferroptose, régule la sensibilité des cellules à cette voie, plutôt que d’induire directement la ferroptose. Il reste à déterminer si p53 régule d’autres gènes impliqués dans la ferroptose (Figure 2).
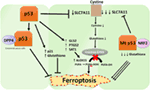
la Figure 2. Les différents rôles de p53 dans la ferroptose., L’Inhibition de la glutathion peroxydase 4 (GPX4), l’enzyme clé qui catalyse la conversion des acides gras polyinsaturés (AGPI) contenant des peroxydes en alcools, est le principal moteur de la ferroptose. Selon le contexte, p53 peut supprimer la ferroptose (comme dans les cellules cancéreuses colorectales) ou favoriser la ferroptose. Le Mutant p53 sensibilise les cellules à la ferroptose encore plus que le p53 de type sauvage.,
dans certaines cellules, p53 régule négativement la Ferroptose
Une étude récemment publiée par Tarangelo et ses collègues montre que p53 régule négativement la ferroptose dans les cellules cancéreuses (50). Ce groupe a constaté que le prétraitement des cellules avec Nutlin-3, un composé qui stabilise p53 retarde l’apparition de la ferroptose dans plusieurs types de cellules. L’apparition retardée de la ferroptose dépend de CDKN1A (codant p21), une cible transcriptionnelle critique de p53., Le mécanisme par lequel p21 retarde la ferroptose n’a pas encore été élucidé, mais on pense que la conservation du glutathion intracellulaire peut être un facteur contribuant à la réduction de la sensibilité à la ferroptose. Les auteurs concluent que l’axe p53–p21 permet aux cellules cancéreuses de survivre dans des conditions de stress métabolique, telles que la privation de cystine, en supprimant l’apparition de la ferroptose (50). Une étude récente a montré que p53 inhibe la ferroptose dans les cellules cancéreuses colorectales en se liant à l’enzyme dipeptidyl-peptidase-4 (DPP4), qui est un modulateur de la ferroptose et du métabolisme lipidique., Mécaniquement, cette étude a montré que la p53 Antagonise la ferroptose en séquestrant la DPP4 dans un pool enzymatique nucléaire inactif. En l’absence de p53, DPP4 est libre d’interagir et de former un complexe avec NOX1; cela conduit à une peroxydation lipidique accrue et à une ferroptose. L’Inhibition de la DPP4 supprime significativement la ferroptose, tandis que la surexpression de la DPP4 déclenche la sensibilité à L’Érastine, en particulier dans les cellules appauvries en p53 (51). Le contrôle bidirectionnel de la ferroptose par p53 par des mécanismes dépendant de la transcription et indépendants de la transcription peut être dépendant du contexte ou du type cellulaire (Figure 2).,
le polymorphisme P47S de TP53 affecte la Ferroptose et la Suppression tumorale
en plus des mutations missense, il existe plusieurs polymorphismes mononucléotidiques (SNP) fonctionnellement significatifs dans le gène TP53 et d’autres protéines connues pour réguler cette voie (telles que MDM2 et MDM4). La variante Pro47Ser (ci-après S47) est le deuxième SNP le plus commun trouvé dans la région de codage p53 (après Pro72Arg) qui modifie la séquence d’acides aminés de la protéine., Pour mieux élucider l’impact de cette variante sur la fonction p53 et le risque de cancer, le groupe de Murphy a généré un modèle de souris humanisée p53 knock-in, dans lequel les exons 4-9 de p53 murin ont été remplacés par des exons humains p53 contenant soit le type sauvage, soit la variante S47 (52-55). La majorité des souris S47 ont spontanément développé des tumeurs de divers types histologiques, en particulier le cancer du foie, entre 12 et 18 mois, contrairement aux souris WT p53 (48)., Dans les fibroblastes embryonnaires de souris et les lignées cellulaires lymphoblastoïdes humaines, la variante S47 a montré une altération de la mort cellulaire programmée en réponse au cisplatine et à d’autres stress génotoxiques. Mécaniquement, la variante S47 est défectueuse pour la transactivation de gènes impliqués dans le métabolisme, tels que Gls2 (glutaminase 2) et Sco2 (48). En accord avec le rôle de Gls2 dans la ferroptose, ce groupe a constaté que les cellules S47 étaient nettement résistantes aux agents induisant la ferroptose Erastin et RSL3 (47, 48). Ce défaut peut contribuer au phénotype à tendance tumorale observé chez les souris S47.,
le Mutant p53 sensibilise les cellules tumorales à la Ferroptose
Le type sauvage p53 régule négativement l’expression de L’importateur de cystine SLC7A11, qui inhibe la sensibilité à la ferroptose (5). Bien que cette régulation se produise dans les cellules normales, dans les cellules tumorales, d’autres médiateurs de SLC7A11 semblent prédominer dans la régulation de ce gène. Par exemple, le facteur de transcription antioxydant principal NRF2 peut également réguler l’expression de SLC7A11 au niveau transcriptionnel, et NRF2 a été impliqué en tant qu’acteur clé dans la protection des cellules cancéreuses contre la ferroptose., Par exemple, l’inhibition de NRF2 dans les cellules cancéreuses hépatocellulaires augmente l’activité anticancéreuse de L’Érastine et du sorafénib in vivo (56). Les formes mutantes de p53 peuvent inhiber la fonction NRF2 par interaction directe, et un groupe a constaté que les tumeurs avec le mutant p53 contiennent de très faibles niveaux de SLC7A11 et montrent ainsi une sensibilité accrue à la ferroptose. Notamment, la surexpression de SLC7A11 dans les modèles Mutants p53 a conduit à une résistance aux médicaments, ce qui suggère que les niveaux d’expression de SLC7A11 doivent être pris en compte lors du ciblage de cancers Mutants entraînés par p53 avec des composés induisant la ferroptose (57)., À l’appui de cette prémisse, des travaux récents sur le cancer colorectal (CRC), où la mutation ou la délétion de p53 est un événement fréquent, ont montré que les lignées cellulaires CRC humaines hébergeant le mutant p53 étaient beaucoup plus sensibles à la mort cellulaire médiée par L’Érastine que les cellules CRC avec WT p53. Pour valider ces résultats, ils ont montré que le choc d’une mutation du point chaud p53 dans les cellules HCT116 et SW48 rétablissait la sensibilité à Erastin (51). Ces données mettent en évidence un nouveau mécanisme par lequel les cancers entraînés par le mutant p53 peuvent être exploités en utilisant une thérapie ciblée.,
Conclusion
le rôle de p53 dans le métabolisme est assez clair et peut-être même intuitivement évident: WT p53 limite le métabolisme du glucose et la synthèse des lipides, tandis que le mutant p53 semble faire le contraire. La contribution de son rôle métabolique à la suppression tumorale par p53, et à la capacité du mutant p53 à conduire la progression tumorale, reste à prouver sans équivoque. Le rôle de p53 dans la régulation de la ferroptose, et la contribution de cette fonction, à la suppression tumorale est encore moins clair., Bien que des données convaincantes provenant de modèles murins soutiennent la prémisse que p53 régule la sensibilité des cellules à la ferroptose, cela peut être limité à la capacité de p53 basal à supprimer le développement tumoral spontané, et dans les modèles murins stressés par l’oncogène, il est clair que la sénescence et l’apoptose jouent le rôle prédominant. De même, p53 peut réguler la sensibilité à la ferroptose d’une manière spécifique au type de cellule. D’autres études sur des modèles animaux, portant sur la ferroptose dans différents tissus, doivent être effectuées pour mieux comprendre le rôle de p53 dans la ferroptose et la ferroptose dans la suppression tumorale., De plus, une idée plus claire de ce que les gènes cibles p53 jouent un rôle dans la sensibilité à la ferroptose doit être atteinte. La résolution de ces questions devrait fournir de nouvelles avenues indispensables pour lutter contre les tumeurs avec le mutant p53.
contributions des auteurs
KG, SB, TB, AB-K, C-PK et MM ont chacun écrit un à deux paragraphes de cet article. KG et SB ont fait le chiffre. KG et MM ont décrit le chapitre.,
déclaration de conflit d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
le réviseur OAF et l’éditeur de traitement ont déclaré leur affiliation commune.
Remerciements
Les recherches présentées dans cette publication ont été appuyées par les National Institutes of Health sous les numéros CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) et T32 CA009171 (TB)., Le contenu est de la seule responsabilité des auteurs et ne représente pas nécessairement les opinions officielles des National Institutes of Health.
3. Humpton TJ, Vousden KH. Régulation du métabolisme cellulaire et de l’hypoxie par p53. Printemps Froid Harb Perspect Med (2016) 6(7):211-30. doi: 10.1101 / cshperspect.a026146
CrossRef Texte Intégral | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et coll., Le suppresseur de tumeur p53 régule négativement la glycolyse stimulée par l’hypoxie par son RRAD cible. Oncotarget (2014) 5(14):5535-46. doi: 10.18632 / oncotarget.2137
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
17. Lee M, Yoon JH. Interaction métabolique entre la glycolyse et l’oxydation mitochondriale: l’effet Warburg inverse et son implication thérapeutique. Monde J Biol Chem (2015) 6(3):148-61. doi: 10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., La déplétion du nouveau gène p53-cible carnitine palmitoyltransférase 1C retarde la croissance tumorale dans le modèle tumoral de type I de la neurofibromatose. La Mort Cellulaire Diffèrent (2013) 20(4):659-68. doi: 10.1038 / cdd.2012.168
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari m, Bradley DR, Gleave ME, et al. Le dérèglement de sterol response element-binding protéines et des effecteurs en aval dans le cancer de la prostate au cours de la progression de l’indépendance de l’androgène., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Les souris frappées avec un gène chimérique humain/murin p53 se développent normalement et montrent des réponses p53 de type sauvage aux agents dommageables à l’ADN: un nouvel outil de recherche biomédicale. Oncogène (2001) 20(3):320-8. doi: 10.1038 / sj.onc.1204080
PubMed Abstract | CrossRef Texte Intégral | Google Scholar
57. Liu DS, Duong CP, Haupt s, Montgomery KG, House CM, Azar WJ, et al. L’inhibition de l’axe système XC-/glutathion cible sélectivement les cancers présentant une accumulation de mutant-p53., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















