Genome Spot (Français)
L’analyse des voies est devenue une procédure si courante en bioinformatique, en particulier dans l’étude de l’expression des gènes. Si vous regardez une enquête sur des articles récents, il semble qu’il y ait un tas de façons de le faire. Dans ce post, je vais discuter des différences entre deux outils couramment utilisés; DAVID et GSEA.

Les concepts derrière les deux algorithmes sont très différents., DAVID détermine les chevauchements entre les listes de gènes fournies par l’utilisateur et les bases de données organisées, à la recherche de chevauchements plus importants que ceux attendus par hasard. Vous pouvez améliorer la précision de l’algorithme en fournissant un fichier d’arrière-plan contenant tous les gènes considérés/détectés dans l’expérience. Les ensembles de gènes fournis par l’utilisateur sont normalement générés en sélectionnant des gènes qui passent un seuil de signification. La procédure DAVID est similaire à d’autres disponibles tels que Ingenuity, AmiGO et GeneGO., La sélection des valeurs de signification est largement arbitraire, mais il est courant de définir le seuil à FDR adjusted p<0.05. La modification du seuil modifiera considérablement les résultats de l’enrichissement. Les tests statistiques utilisés comprennent hypergéométrique, exact de Fisher et Chi-carré.
Contrairement aux méthodes ci-dessus qui nécessitent des listes de gènes qui passent des seuils arbitraires, GSEA est un outil qui utilise chaque point de données dans son algorithme statistique. Dans la méthode « classique », les gènes sont classés de la plus régulée vers le haut à la plus régulée vers le bas., La mesure de rang elle-même varie, mais deux méthodes valides consistent à utiliser la valeur p signée ou un intervalle de confiance inférieur à 90% du changement de pli. La base du test est d’évaluer si les membres d’un ensemble de gènes semblent enrichis à une extrémité du profil. Pour tester l’enrichissement, GSEA effectue des permutations du profil, en calculant l’enrichissement du gène défini mille fois ou plus pour estimer empiriquement les valeurs p. D’autres outils qui utilisent l’ensemble du profil effectuent des tests analytiques comme le test de Wilcoxon et de Kolmogorov-Smirnov.,
Donc maintenant que nous comprenons les différences principales dans les algorithmes, nous pouvons commencer à comparer les résultats de ces deux techniques. J’ai revisité un ensemble de données dont j’ai déjà parlé, le traitement à l’azacitidine dans les cellules AML3 profilées par ARN-seq. Je vais utiliser les données d’expression différentielle ARN-seq générées par DESeq comme point de départ pour comparer DAVID et GSEA.
pour DAVID, j’ai fait un fichier d’arrière-plan composé de tous les gènes 19,030 détectés dans l’expérience au-dessus du seuil de détection. Le significatif (FDR <0.,05) les listes de gènes dans la Direction up (2502 gènes) et down (2907 gènes) ont été soumises séparément à DAVID 6.7 pour analyse, en se concentrant uniquement sur les voies de KEGG.
Pour GSEA, j’ai utilisé la même procédure métrique de rang décrite précédemment. J’ai utilisé l’algorithme GSEA classique avec le gsea2-2.2.2.exécutable jar, utilisant les ensembles de gènes KEGG de MSigDB v5.1.
dans le graphique ci-dessous, j’ai déterminé le chevauchement des ensembles de gènes statistiquement significatifs (FDR<0.05) séparément pour les voies régulées vers le haut et vers le bas., J’ai également inclus une intersection des résultats de DAVID lorsqu’aucun arrière-plan n’est fourni (pour la curiosité seulement, pas une procédure recommandée).
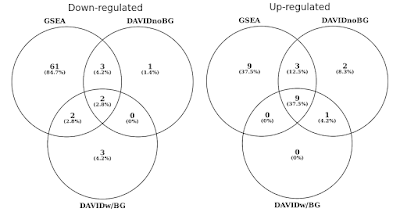
DAVID vs GSEA en utilisant le même profil d’expression génique. Les nombres représentent des ensembles de gènes qui sont significativement enrichis (FDR<0.05).
Vous remarquerez que dans les deux sens réglementé vers le haut et vers le bas, GSEA identifie des résultats plus significatifs par rapport à DAVID. J’ai trouvé que c’était également le cas dans d’autres bibliothèques d’ensembles de gènes telles que REACTOME et GO., La différence de sensibilité entre les deux outils se résume aux différents algorithmes, DAVID utilisant une liste discrète de gènes, par rapport à GSEA qui utilise chaque point de données. GSEA sera beaucoup plus sensible dans l’identification des ensembles de gènes qui ont de nombreux membres qui subissent des changements subtils dans l’expression. Individuellement, ces changements ne sont pas statistiquement significatifs, mais lorsqu’une grande fraction des gènes d’un ensemble change collectivement d’expression, cette tendance pourrait être très significative statistiquement parlant.,
En plus de l’algorithme plus sensible, les ensembles de gènes de DAVID et les annotations de gènes sont vraiment obsolètes. La dernière mise à jour des nombreuses sources de données de DAVID Date du milieu à la fin de 2009. En tant que tel, de nombreuses accessions de gènes D’Ensembl peuvent ne pas être prises en charge. En effet, de mon dossier de fond qui contenait 19 030 seulement 13 488 ont été reconnus par DAVID. Le manque d’ensembles de gènes mis à jour est également une préoccupation, tout comme le nombre toujours croissant de citations que DAVID reçoit encore.,
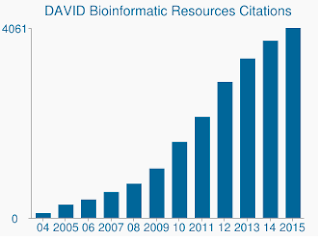
DAVID reçoit toujours des milliers de citations par an malgré les données sous-jacentes qui sont terriblement obsolètes.
En conclusion, je recommanderais aux chercheurs d’utiliser des outils qui examinent un profil entier pour déterminer l’enrichissement (GSEA, CAMERA, WilcoxGST, Pathifier) plutôt que de soumettre des ensembles de gènes pour l’analyse de chevauchement. L’utilisation de DAVID pourrait être l’option rapide et facile, mais cela se fait au détriment d’une réduction massive de la sensibilité.
Pour en savoir plus
JA Timmons, KJ Szkop, IJ Gallagher., De multiples sources de biais confondent l’analyse d’enrichissement fonctionnel des données omiques globales. Biologie du génome, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. L’Impact de l’accumulation des connaissances sur la voie de l’enrichissement de l’analyse. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: en mai 2016, DAVID a été mis à jour!
https://david.ncifcrf.gov/content.jsp?file=release.html















