genoom Spot
Pathway analyse is zo ‘ n veel voorkomende procedure geworden in de bioinformatica, vooral bij het bestuderen van genexpressie. Als je kijkt naar een overzicht van recente papers het lijkt erop dat er een heleboel manieren waarop het kan worden gedaan. In deze post, Ik zal de verschillen tussen twee veelgebruikte tools te bespreken; DAVID en GSEA.

de concepten achter de twee algoritmen zijn zeer verschillend., DAVID bepaalt overlappingen tussen door de gebruiker geleverde genenlijsten en de gecureerde databases, op zoek naar overlappingen die groter zijn dan die verwacht door willekeurige toeval. U kunt de nauwkeurigheid van het algoritme verbeteren door een achtergrondbestand te verstrekken dat alle genen bevat die in het experiment werden overwogen/gedetecteerd. De door de gebruiker geleverde genreeksen worden normaal geproduceerd door genen te selecteren die een significantiedrempel passeren. De DAVID procedure is vergelijkbaar met andere beschikbare zoals vindingrijkheid, AmiGO en GeneGO., De selectie van significantiewaarden is grotendeels willekeurig, maar het is gebruikelijk om de drempelwaarde in te stellen op FDR gecorrigeerde p<0,05. Het wijzigen van de drempel zal de verrijkingsresultaten aanzienlijk veranderen. De gebruikte statistische tests omvatten hypergeometrisch, Fisher ‘ s exact en Chi-kwadraat.
In tegenstelling tot de bovenstaande methoden die lijsten van genen vereisen die willekeurige drempels passeren, is GSEA een hulpmiddel dat elk datapoint in zijn statistische algoritme gebruikt. In de” klassieke ” methode worden de genen gerangschikt door van het meest up-geregeld aan het meest down-geregeld., De rangmetriek zelf varieert, maar twee geldige methoden zijn om de ondertekende p-waarde te gebruiken, of een lager 90% betrouwbaarheidsinterval van de vouwverandering. De basis van de test is om te beoordelen of leden van een genset verrijkt lijken aan één uiteinde van het profiel. Om de verrijking te testen, voert GSEA permutaties van het profiel uit, waarbij de verrijking van het gen duizend of meer keren wordt berekend om p-waarden empirisch te schatten. Andere tools die het volledige profiel gebruiken voeren analytische tests uit zoals de Wilcoxon-en Kolmogorov-Smirnov-test., nu we de belangrijkste verschillen in de algoritmen begrijpen, kunnen we beginnen met het vergelijken van de resultaten van deze twee technieken. Ik revisited een dataset ik heb eerder besproken, azacitidine behandeling in aml3 cellen geprofileerd door RNA-seq. Ik zal de differentiële RNA-seq uitdrukkingsgegevens gebruiken die door deseq als beginpunt worden geproduceerd om DAVID en GSEA te vergelijken.
voor DAVID heb ik een achtergrondbestand gemaakt dat bestaat uit alle 19.030 genen die in het experiment boven de detectiedrempel zijn gedetecteerd. De significante (FDR<0.,05) genlijsten in de richting omhoog (2502 genen) en omlaag (2907 genen) werden afzonderlijk voorgelegd aan DAVID 6.7 voor analyse, gericht op Kegg paden alleen.
voor GSEA gebruikte ik dezelfde rank metric procedure als eerder beschreven. Ik gebruikte het klassieke gsea algoritme met de gsea2-2.2.2.jar uitvoerbaar, met behulp van Kegg gen sets van MSigDB v5. 1.
in de onderstaande grafiek bepaalde ik de overlap van statistisch significante genensets (FDR<0,05) afzonderlijk voor op en neer gereguleerde routes., Ik heb ook een kruising van DAVID resultaten opgenomen wanneer er geen achtergrond wordt verstrekt (alleen voor nieuwsgierigheid, niet een aanbevolen procedure).
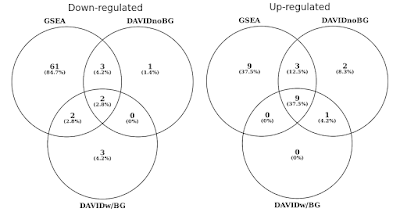
DAVID vs GSEA met hetzelfde genexpressieprofiel. Getallen vertegenwoordigen genre sets die significant verrijkt zijn (FDR<0,05).
U zult merken dat in zowel de op-als neer geregelde richting, gsea meer significante resultaten identificeert in vergelijking met DAVID. Ik vond dit was ook het geval in andere gen set bibliotheken zoals REACTOME en GO., Het verschil in gevoeligheid tussen de twee tools komt neer op de verschillende algoritmen, met DAVID met behulp van een discrete lijst van genen, versus GSEA die elk gegevenspunt gebruikt. GSEA zal veel gevoeliger zijn in het identificeren van genre sets die veel leden hebben die subtiele veranderingen in expressie ondergaan. Individueel zijn deze veranderingen niet statistisch significant, maar wanneer een groot deel van genen in een reeks collectief verschuift in expressie, kan die trend statistisch gezien zeer significant zijn.,
naast het gevoeligere algoritme zijn Davids genverzamelingen en genannotaties echt verouderd. De laatste update van DAVID ‘ s vele gegevensbronnen zijn midden tot eind 2009. Als zodanig kunnen veel genentoetredingen uit Ensembl niet worden ondersteund. Inderdaad, uit mijn achtergrondbestand dat 19.030 bevatte, werden slechts 13.488 door DAVID herkend. Het ontbreken van bijgewerkte genensets is ook een punt van zorg, evenals het steeds groeiende aantal citaties dat DAVID nog steeds ontvangt.,
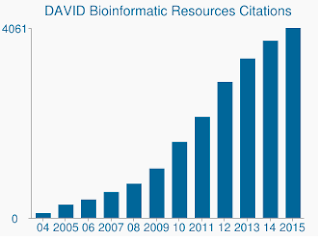
DAVID ontvangt nog steeds duizenden citaties per jaar, ondanks dat de onderliggende gegevens helaas verouderd zijn.
concluderend zou ik onderzoekers aanraden tools te gebruiken die naar een volledig profiel kijken om verrijking te bepalen (GSEA, CAMERA, WilcoxGST, Pathifier) in plaats van Gensets voor overlapanalyse in te dienen. Het gebruik van DAVID is misschien de snelle en gemakkelijke optie, maar dit gaat ten koste van een enorme vermindering van de gevoeligheid.JA Timmons, KJ Szkop, IJ Gallagher., Meerdere bronnen van bias verwarren functionele verrijkingsanalyse van global-omics gegevens. Genome biology, 2015. Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Impact van kennisaccumulatie op de analyse van pathway enrichment. bioRxiv. doi: http://dx.doi.org/10.1101/049288
bewerken: in Mei 2016 werd DAVID bijgewerkt!
https://david.ncifcrf.gov/content.jsp?file=release.html















