Frontiers in Endocrinology (Polski)
wprowadzenie
Gen suppressor TP53 jest najbardziej zbadanym ludzkim genem od czasu jego odkrycia prawie 40 lat temu (1). Głównym powodem tego statusu jest krytyczna rola p53 w zapobieganiu rozwojowi raka i jest powszechnie uważany za ” strażnika genomu.,”Od pewnego czasu powszechnie uważa się, że rola p53 w supresji guza wynika z jego zdolności do indukowania apoptozy, zatrzymania cyklu komórkowego i starzenia się komórek przedrakowych (2). Jednak teraz jest coraz bardziej jasne, że p53 reguluje wiele innych szlaków w komórce i że te inne szlaki również odgrywają rolę w zdolności p53 do działania jako supresor nowotworu (3). W szczególności rola p53 w regulacji genów biorących udział w metabolizmie i ferroptozie została zaangażowana w jego zdolność do hamowania rozwoju nowotworu., Ferroptosis jest nową ścieżką śmierci komórki po raz pierwszy scharakteryzowaną w 2012 roku i może być najlepiej opisana jako zależna od żelaza, niezależna od kaspazy forma śmierci komórki napędzana przez powstawanie peroksydacji lipidów (4). W szczególności dwa mysie modele zawierające zmodyfikowane mutacje w p53, które eliminują zdolność p53 do indukowania apoptozy i starzenia, oba zachowują zdolność do tłumienia spontanicznego rozwoju nowotworu; oba te mutanty zachowują zdolność do transactivate genów w metabolizmie i ferroptosis (5, 6)., Poniżej przedstawiono podsumowanie danych dotyczących wpływu p53 na regulację metabolizmu i ferroptozy.
Wild-Type (WT) p53 pozytywnie reguluje fosforylację oksydacyjną i hamuje metabolizm glukozy
Wild-type p53 reguluje wszechstronność metaboliczną komórek poprzez faworyzowanie oddychania mitochondrialnego nad glikolizą, częściowo poprzez transaktywację SCO2 (zespołu oksydazy cytochromu c), która odgrywa bezpośrednią rolę w fosforylacji oksydacyjnej (7)., p53 reguluje również bezpośrednio transaktywację GLS2 (Glutaminazy 2); enzym ten umożliwia wykorzystanie glutaminy jako źródła energii dla mitochondriów (8). Ponadto WT P53 negatywnie reguluje glikolizę poprzez transkrypcyjne hamowanie transporterów glukozy GLUT1 i GLUT4 oraz przez transactivating RRAD i TIGAR; oba są inhibitorami glikolizy (9-11). Wreszcie, p53 również bezpośrednio wiąże się i hamuje enzym dehydrogenazy glukozo-6-fosforanowej, hamując w ten sposób metabolizm glukozy (12)., Z tych i innych badań wynika, że w normalnych, nieakcentowanych organizmach p53 bezpośrednio reguluje stan metaboliczny w komórce (ryc. 1). Nic dziwnego, że gen ten i wiele jego regulatorów jest zaangażowanych w Choroby metaboliczne, w tym otyłość i cukrzycę (13).
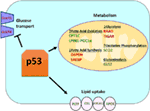
Rysunek 1. Rola typu dzikiego (WT) p53 w metabolizmie. Geny pozytywnie regulowane przez p53 są pokazane na zielono, a geny negatywnie regulowane przez p53 są pokazane na Czerwono., p53 hamuje transport glukozy, glikolizę i syntezę kwasów tłuszczowych, podczas gdy Promuje wychwyt lipidów, utlenianie kwasów tłuszczowych, fosforylację oksydacyjną i glutaminolizę.
zmutowany p53 pozytywnie reguluje metabolizm Warburga (glikoliza tlenowa)
w przeciwieństwie do funkcji WT P53, zmutowany p53 w komórkach nowotworowych sprzyja glikolizie tlenowej, częściowo poprzez zwiększenie przepływu transportera glukozy GLUT1 do błony osocza, co zwiększa import glukozy (14, 15)., Po mutacji p53 zmniejszone poziomy SCO2 i GLS2 oraz zwiększone poziomy GLUT1 i GLUT4 sprzyjają glikolizie tlenowej nad fosforylacją oksydacyjną. W ten sposób uważa się, że zmutowany p53 przyczynia się do skłonności komórek nowotworowych do wykorzystania glikolizy tlenowej na rzecz fosforylacji oksydacyjnej lub tak zwanego metabolizmu Warburga (15). Jedną z cech rozpoznawczych raka jest deregulowany metabolizm, na ogół wykazywany przez przejście z glikolizy tlenowej na fosforylację oksydacyjną., Chociaż powoduje to mniejszą i mniej wydajną wydajność ATP, uważa się, że komórki nowotworowe korzystają z odwracania glikolitycznych półproduktów do szlaków biosyntetycznych niezbędnych do szybkiego podziału komórek (16). Ta zmiana metaboliczna prowadzi również do zmniejszonej apoptozy mitochondriów i bardziej wydajnej sygnalizacji poprzez dostępne metabolity w komórkach nowotworowych (17).
wspólny wariant genetyczny w TP53 wpływa na jego funkcję w metabolizmie
istnieje wspólny polimorfizm regionu kodującego p53 w kodonie 72, kodujący dla proliny (P72) lub argininy (R72)., Ta zmienność aminokwasów może wpływać na funkcję p53 w odniesieniu do losu komórek po stresie. W odpowiedzi na uszkodzenie DNA, wariant P72 p53 wywołuje głównie zatrzymanie cyklu komórkowego, podczas gdy wariant R72 indukuje głównie śmierć komórki lub apoptozę (18, 19). Pomimo tych różnic w funkcji, zmienność kodonu 72 nie była konsekwentnie związana z podatnością na raka (20). Natomiast w badaniach na ludziach ten polimorfizm jest znacząco związany ze zwiększonym wskaźnikiem masy ciała i ryzykiem cukrzycy (21, 22)., Założenie to jest poparte badaniami na myszach, w których model myszy dla tych wariantów kodonu 72 wykazuje zwiększoną cukrzycę wywołaną dietą wysokotłuszczową u myszy z wariantem R72, w porównaniu do P72. W badaniach tych geny docelowe p53 TNFa i Npc1l1 zostały zidentyfikowane jako kluczowe Regulatory wzrostu otyłości indukowanej dietą u myszy z R72 (23). Co ciekawe, wykazano również, że wariant R72 zapewnia zwiększone przeżycie komórek w odpowiedzi na deprywację składników odżywczych (24)., Wyniki te doprowadziły do hipotezy, że wariant R72 p53 powstał i został wybrany jako populacje migrujące na północ, gdzie zimna pogoda wymagałaby zwiększonej akumulacji tłuszczu, ale gdzie przetrwanie w odpowiedzi na deprywację składników odżywczych również byłoby pod selekcją (24).
p53 reguluje metabolizm lipidów
chociaż p53 jest dobrze znany z regulacji glikolizy i cyklu kwasu cytrynowego, wykazano, że p53 odgrywa również rolę w regulacji metabolizmu lipidów (25)., Uważa się, że WT p53 zwiększa utlenianie kwasów tłuszczowych, hamując syntezę kwasów tłuszczowych, działając w ten sposób jako ujemny regulator syntezy lipidów (25). Istnieje kilka genów docelowych p53 z rolą w metabolizmie lipidów. Sanchez-Macedo i współpracownicy wykazali, że palmitoilotransferaza karnityny 1C (CPT1C) jest transkrypcyjnie regulowana przez p53; enzym ten pomaga w transporcie aktywowanych kwasów tłuszczowych do mitochondriów., W celu wsparcia roli tego genu regulowanego przez p53 w raku, grupa ta wykazała, że myszy z niedoborem Cpt1c wykazują opóźniony rozwój guza i wyższe wskaźniki przeżycia (26). Lipina 1 (LPIN1) jest kolejnym genem docelowym p53; LPIN1 jest niezbędny do prawidłowego rozwoju adipocytów i jest indukowany w warunkach niskiej zawartości składników odżywczych (27). Finck i współpracownicy wykazali, że LPIN1 oddziałuje z PGC-1α, innym znanym genem docelowym p53, który odgrywa rolę w metabolizmie, i że interakcja ta aktywuje ekspresję genów zaangażowanych w promowanie utleniania kwasów tłuszczowych (28).,
oprócz bezpośredniej regulacji transkrypcji genów zaangażowanych w metabolizm lipidów, p53 może również regulować metabolizm lipidów w sposób obejmujący bezpośrednią interakcję białko–białko. Na przykład dehydrogenaza glukozo-6-fosforanowa, która jest enzymem ograniczającym szybkość w szlaku pentozofosforanowym, wiąże się i jest bezpośrednio hamowana przez p53, powodując zmniejszenie produkcji NADPH i w konsekwencji zmniejszenie syntezy kwasów tłuszczowych (12)., Rodzina czynników transkrypcyjnych steroli regulatory element-binding proteins (SREBP) moduluje ekspresję genów biorących udział w syntezie cholesterolu, kwasów tłuszczowych, triacyloglicerolu i fosfolipidów (29-31). WT p53 hamuje funkcję SREBP (32), podczas gdy zmutowane formy p53 wiążą się bezpośrednio ze SREBP i zwiększają ich funkcję transkrypcyjną, co prowadzi do zwiększenia aktywności SREBP w guzach ludzkich (33, 34). W konsekwencji zmutowany p53 jest skorelowany z wyższą ekspresją genów biosyntezy steroli w guzach piersi u ludzi (34, 35)., Wreszcie, kinaza białkowa aktywowana AMP (AMPK) jest enzymem aktywowanym pod niskim poziomem składników odżywczych lub stresem energetycznym i wiadomo, że hamuje syntezę kwasów tłuszczowych poprzez interakcję z acetylo-CoA-karboksylazą i SREBP-1 (36, 37). Zhou i współpracownicy wykazali, że zmutowany p53 preferencyjnie wiąże się i hamuje AMPK, co prowadzi do zwiększenia syntezy kwasów tłuszczowych. W rezultacie zmutowane białka p53 prowadzą do zwiększonej sygnalizacji AMPK, przyczyniając się do inwazyjnego wzrostu komórek nowotworowych (33). Mniej zbadanym obszarem jest rola p53 w transporcie lipidów., Wykazano, że p53 transkrypcyjnie reguluje apolipoproteinę B (apoB) i Kompleks enzymu edytującego apoB 1, wskazując na rolę p53 w regulacji miażdżycowych lipoprotein (38). Analiza Microarray ludzkich komórek pochodzących z wątroby zidentyfikowała białko transferu fosfolipidów, kasetę wiążącą ATP A12 i lipazę karboksylową jako trzy geny docelowe p53, które odgrywają rolę w transporcie lipidów(39, 40)., Ogólnie rzecz biorąc, chociaż oczywiste jest, że p53 odgrywa kluczową rolę w mediacji syntezy lipidów i metabolizmu, udział tego szlaku, a te geny docelowe p53, do supresji guza przez p53 pozostaje do ustalenia (Rysunek 1).
Ferroptosis jest nowatorskim szlakiem śmierci komórek napędzanym peroksydacją lipidów
w 2012 roku Dixon i współpracownicy odkryli nową formę regulowanej śmierci komórek o nazwie ferroptosis. Ferroptoza jest zależną od żelaza, niezależną od kaspazy formą śmierci komórki w wyniku nagromadzenia utlenionych lipidów (4, 41)., Proces ten jest napędzany przez inaktywację peroksydazy glutationowej 4 (GPX4), enzymu odpowiedzialnego za konwersję śmiertelnych hydroperoksydów lipidowych do nietoksycznych alkoholi lipidowych, które wymagają glutationu do funkcjonowania (41). Uważa się, że peroksydacja wielonienasyconych kwasów tłuszczowych (pufa) jest impulsem do śmierci komórek przez ferroptozę. Pufa zawierają protony bis-allilowe, które mogą być łatwo absorbowane i wytwarzają rodniki, które będą reagować z tlenem, tworząc więcej rodników i powodując reakcję łańcuchową reaktywnych lipidowych form tlenu (42)., Dokładny mechanizm śmierci komórki przez ferroptozę pozostaje nieznany, ale jedna z hipotez mówi, że uszkodzenie lipidów prowadzi do zniszczenia błony plazmowej (43). Spekulowano, że ferroptosis może być mechanizmem supresji nowotworu, który działa poprzez eliminację komórek, które są pozbawione składników odżywczych lub były narażone na stres środowiskowy lub infekcję.,
farmakologiczna Regulacja Ferroptozy
może być wywołana za pomocą inhibitorów układu xc− takich jak erastyna, lub analogów, takich jak glutaminian i sorafenib, które hamują import cystyny, powodując zubożony glutation i późniejszą inaktywację GPX4. Alternatywnie, ferroptoza może być wywołana przez (1S,3R)-RSL3 (dalej zwane RSL3), który bezpośrednio wiąże się i hamuje GPX4 (4, 5, 42). Ale sulfoksymina, FIN56, FINO2, CCl4 i cisplatyna są inne czynniki, które zostały wykazane, aby wywołać ferroptosis w komórkach., Śmierć przez ferroptosis można zapobiec przez tłumienie peroksydacji lipidów, które mogą być osiągnięte za pomocą lipofilowych przeciwutleniaczy, takich jak ferrostatin-1, liproxstatin-1, lub witamina E. chelatory żelaza, takie jak deferoksamina lub cicloprox są innym narzędziem stosowanym do tłumienia ferroptosis poprzez zmniejszenie poziomu żelaza. Zubożenie Pufa lub dodanie jednonienasyconych kwasów tłuszczowych do pożywek hodowli komórkowej może również uratować komórki przed ferroptozą (42, 44).,
Ferroptosis jest zaangażowany w supresję guza za pośrednictwem P53
w 2012 roku Gu i współpracownicy opracowali model myszy, w którym trzy normalnie acetylowane pozostałości lizyny w domenie wiążącej DNA p53 zostały zmutowane do argininy, a zatem nie mogły być acetylowane; ta mysz jest określana jako mysz 3kR. W szczególności komórki myszy 3KR nie są w stanie poddać się apoptozie zależnej od p53, zatrzymaniu cyklu komórkowego lub starzeniu, i rzeczywiście mutant 3kR p53 nie przenosi większości genów docelowych p53., Co ciekawe, ten mysi model nie rozwija się samoistnie, co sugeruje, że p53 może hamować rozwój guza niezależnie od starzenia się lub apoptozy (45). Grupa ta odkryła, że zmutowane białko 3kR zachowuje zdolność do ulegania ferroptozie i regulowania metabolizmu cystyny poprzez regulację ekspresji importera cystyny SLC7A11; sugerowało to, że ferroptoza może być jedną ze ścieżek, która leży u podstaw supresji guza za pośrednictwem p53., Kiedy MEFS typu dzikiego i MEFS 3KR były leczone induktorem ferroptozy Erastyn, zaobserwowano prawie 50% śmierci komórek, podczas gdy MEFS typu null p53 wykazywały 20% śmierci komórek; wskazuje to, że p53 uczula komórki na ferroptozę, a także, że inne kluczowe regulatory również odgrywają rolę w ferroptozie (5). Następnie gu i współpracownicy zidentyfikowali dodatkowe miejsce acetylacji w lizynie 98 p53 i wygenerowali model myszy, w którym wszystkie cztery miejsca acetylacji zostały zmutowane do argininy (4KR)., Co ciekawe, mutant 4KR nie był w stanie regulować genów zaangażowanych w ferroptosis jak SLC7A11, i w przeciwieństwie do mutanta 3kR nie był w stanie zahamować rozwoju nowotworu (46). Choć obecnie korelacyjne, dane te implikują rolę p53 w ferroptosis w jego zdolności do tłumienia rozwoju nowotworu.
w komórkach nie przekształconych P53 pozytywnie reguluje Ferroptozę
oprócz SLC7A11 odkryto, że kilka innych genów docelowych p53 odgrywa rolę w ferroptozie. Należą do nich GLS2, PTGS2 i SAT1., Badania z dwóch odrębnych grup potwierdzają rolę GLS2 w ferroptozie, o której wiadomo, że zmniejsza glutation i zwiększa komórkowy poziom ROS. Jiang i współpracownicy stosowali inhibitory ferroptozy w połączeniu z inhibitorami glutaminolizy w celu zahamowania ferroptozy indukowanej Erastyną, wykazując w ten sposób, że ferroptoza wymaga glutaminolizy i GLS2 (47). Murphy i współpracownicy wykazali, że polimorficzny wariant p53 był w stanie wywołać zatrzymanie wzrostu i starzenie się zarówno w ludzkich, jak i mysich komórkach, ale nie udało się zahamować SLC7A11 lub transactivate GLS2., Wariant ten był znacznie osłabiony w indukowaniu ferroptozy i hamowaniu rozwoju guza, tym samym ponownie implikując rolę p53 w supresji guza za pośrednictwem ferroptozy (48). Innym genem docelowym p53 odgrywającym rolę w ferroptozie jest PTGS2, gen kodujący enzym cyklooksygenaza-2. Stockwell i współpracownicy po raz pierwszy wykazali, że indukcja ferroptozy za pomocą Erastyny i RSL3 doprowadziła do zwiększenia liczby PTGS2 (41). W szczególności, PTGS2 nie był regulowany przez induktory ferroptozy w komórkach p53-null, co sugeruje, że regulacja ta jest zależna od p53 (5)., Obecnie wzrost PTGS2 jest szeroko stosowany jako marker ferroptozy (5, 41).
ostatnie badania przeprowadzone przez grupę Gu wykazały, że Gen docelowy P53 SAT1 reguluje ferroptozę (49). Autorzy zidentyfikowali SAT1 jako bezpośredni cel p53 i wykazali, że wyciszanie SAT1 zmniejsza śmierć komórek wywołaną reaktywnymi formami tlenu w komórkach z WT p53, ale nie miało wpływu na komórki p53-null. Mechanicznie grupa ta wykazała, że SAT1 zwiększa poziom i aktywność arachidonianu 15-lipoksygenazy, enzymu wiążącego żelazo, który utlenia Pufa i zwiększa peroksydację lipidów., W szczególności badanie to wykazało, że ani sam p53, ani SAT1 nie wydają się być wystarczające do wywołania ferroptozy. Zamiast tego połączone dane są bardziej zgodne z założeniem, że p53, dzięki regulacji genów, które przyczyniają się do ferroptozy, reguluje wrażliwość komórek na ten szlak, zamiast bezpośrednio indukuje ferroptozę. Czy p53 reguluje inne geny zaangażowane w ferroptozę pozostaje do ustalenia (ryc. 2).
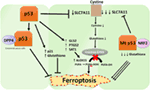
Rysunek 2. Różne role p53 w ferroptozie., Hamowanie peroksydazy glutationowej 4 (GPX4), kluczowego enzymu katalizującego konwersję wielonienasyconych kwasów tłuszczowych (Pufa) zawierających nadtlenki do alkoholi, jest kluczowym czynnikiem ferroptozy. W zależności od kontekstu p53 może hamować ferroptozę (np. w komórkach raka jelita grubego) lub promować ferroptozę. Zmutowany p53 uwrażliwia komórki na ferroptozę nawet bardziej niż dziki p53.,
w niektórych komórkach, P53 negatywnie reguluje Ferroptosis
badanie niedawno opublikowane przez Tarangelo i współpracowników pokazuje, że p53 negatywnie reguluje ferroptosis w komórkach nowotworowych (50). Grupa ta odkryła, że wstępne leczenie komórek Nutlin-3, Związkiem stabilizującym p53 opóźnia początek ferroptozy w kilku typach komórek. Stwierdzono, że opóźniony początek ferroptozy zależy od CDKN1A (kodowanie p21), krytycznego celu transkrypcyjnego p53., Mechanizm, za pomocą którego p21 opóźnia ferroptozę, nie został jeszcze wyjaśniony, ale uważa się, że zachowanie wewnątrzkomórkowego glutationu może być czynnikiem zmniejszającym wrażliwość na ferroptozę. Autorzy wnioskują, że oś p53-p21 umożliwia komórkom nowotworowym przetrwanie w warunkach stresu metabolicznego, takich jak pozbawienie cystyny, poprzez hamowanie początku ferroptozy (50). Ostatnie badania wykazały, że p53 hamuje ferroptozę w komórkach raka jelita grubego przez wiązanie się z enzymem dipeptydylopeptydazy-4 (DPP4), który jest modulatorem ferroptozy i metabolizmu lipidów., Mechanicznie badanie to wykazało, że p53 antagonizuje ferroptozę poprzez sekwestrowanie DPP4 w nieaktywnej puli enzymatycznej jądrowej. W przypadku braku p53, DPP4 może swobodnie wchodzić w interakcje i tworzyć kompleks z NOX1; prowadzi to do zwiększonej peroksydacji lipidów i ferroptozy. Hamowanie DPP4 znacząco hamuje ferroptozę, podczas gdy nadmierna ekspresja DPP4 wyzwala wrażliwość na Eratynę, szczególnie w komórkach zubożonych p53 (51). Dwukierunkowa Kontrola ferroptozy przez p53 poprzez mechanizmy zależne od transkrypcji i niezależne od transkrypcji może być zależna od kontekstu lub typu komórkowego (ryc. 2).,
polimorfizm P47S TP53 wpływa na Ferroptozę i supresję guza
oprócz mutacji missense, istnieje kilka funkcjonalnie znaczących polimorfizmów jednonukleotydowych (SNP) w genie TP53 i innych białkach regulujących ten szlak (takich jak MDM2 i MDM4). Wariant Pro47Ser (zwany dalej S47) jest drugim najczęściej występującym SNP w regionie kodującym p53 (po Pro72Arg), który zmienia sekwencję aminokwasów białka., Aby lepiej wyjaśnić wpływ tego wariantu na funkcję p53 i ryzyko zachorowania na raka, Grupa Murphy ' ego stworzyła humanizowany model p53 knock-in Mysie, w którym exony 4-9 mysich p53 zostały zastąpione przez ludzkie egzony p53 zawierające Typ dziki lub wariant S47 (52-55). Większość myszy S47 spontanicznie rozwinęła nowotwory różnych typów histologicznych, zwłaszcza raka wątroby, między 12 A 18 miesiącem życia, w przeciwieństwie do myszy WT p53 (48)., W przypadku fibroblastów zarodkowych myszy i ludzkich limfoblastoidalnych linii komórkowych wariant S47 wykazał upośledzenie programowanej śmierci komórki w odpowiedzi na cisplatynę i inne naprężenia genotoksyczne. Mechanicznie wariant S47 jest wadliwy do transaktywacji genów biorących udział w metabolizmie, takich jak Gls2 (glutaminaza 2) i Sco2 (48). Zgodnie z rolą Gls2 w ferroptozie, grupa ta wykazała, że komórki S47 były znacznie oporne na środki indukujące ferroptozę Erastyn i RSL3 (47, 48). Wada ta może przyczynić się do fenotypu podatnego na nowotwory obserwowanego u myszy S47.,
zmutowany p53 uwrażliwia komórki nowotworowe na Ferroptozę
dziki Typ p53 negatywnie reguluje ekspresję cystyny SLC7A11, która hamuje wrażliwość na ferroptozę (5). Chociaż regulacja ta występuje w normalnych komórkach, w komórkach nowotworowych, inne mediatory SLC7A11 wydają się dominować w regulacji tego genu. Na przykład główny antyoksydacyjny czynnik transkrypcyjny NRF2 może również regulować ekspresję SLC7A11 na poziomie transkrypcyjnym, a NRF2 został zaangażowany jako kluczowy gracz w ochronie komórek nowotworowych przed ferroptozą., Na przykład hamowanie aktywności NRF2 w komórkach raka wątrobowokomórkowego zwiększa aktywność przeciwnowotworową Erastyny i sorafenibu in vivo (56). Zmutowane formy p53 mogą hamować funkcję NRF2 poprzez bezpośrednią interakcję, a jedna z grup stwierdziła, że guzy z zmutowanym p53 zawierają bardzo niski poziom SLC7A11, a tym samym wykazują zwiększoną wrażliwość na ferroptozę. W szczególności, nadmierna ekspresja SLC7A11 w zmutowanych modelach p53 doprowadziła do oporności na leki, co sugeruje, że poziomy ekspresji SLC7A11 muszą być brane pod uwagę podczas celowania w nowotwory napędzane zmutowanymi p53 związkami indukującymi ferroptozę (57)., Na poparcie tego założenia, niedawne prace w raku jelita grubego (CRC), gdzie mutacja lub delecja p53 jest częstym zdarzeniem, wykazały, że ludzkie linie komórkowe CRC z mutacją p53 były znacznie bardziej wrażliwe na śmierć komórkową za pośrednictwem Erastyny w porównaniu do komórek CRC z WT p53. Aby potwierdzić te wyniki, wykazały, że knock In of a p53 hotspot mutation in both HCT116 and SW48 cells Recovery sensitivity to Erastyn (51). Dane te podkreślają nowatorski mechanizm, za pomocą którego nowotwory napędzane mutantem p53 mogą być wykorzystywane przy użyciu terapii celowanej.,
wniosek
rola p53 w metabolizmie jest dość jasna, a być może nawet intuicyjnie oczywista: WT P53 ogranicza metabolizm glukozy i syntezę lipidów, podczas gdy zmutowany p53 wydaje się działać odwrotnie. Udział jego metabolicznej roli w supresji guza przez p53 oraz w zdolności mutanta p53 do kierowania postępem nowotworu pozostaje do jednoznacznego udowodnienia. Rola p53 w regulacji ferroptozy i udział tej funkcji w supresji guza jest jeszcze mniej wyraźna., Chociaż przekonujące dane z modeli myszy wspierają założenie, że p53 reguluje wrażliwość komórek na ferroptozę, może to być ograniczone do zdolności podstawnej p53 do tłumienia spontanicznego rozwoju nowotworu, a w modelach myszy zestresowanych onkogenem, jasne jest, że starzenie się i apoptoza odgrywają dominującą rolę. Podobnie, p53 może regulować czułość ferroptosis w sposób specyficzny dla typu komórki. Aby lepiej zrozumieć rolę p53 w ferroptozie i ferroptozie w supresji nowotworu, należy przeprowadzić więcej badań na modelach zwierzęcych, z uwzględnieniem ferroptozy w różnych tkankach., Dodatkowo, należy uzyskać jaśniejsze pojęcie o tym, jakie geny docelowe p53 odgrywają rolę w wrażliwości na ferroptozę. Rozwiązanie tych pytań powinno zapewnić bardzo potrzebne nowe możliwości zwalczania guzów z mutantem p53.
wkład autora
KG, SB, TB, AB-K, C-PK i MM każdy napisał jeden do dwóch akapitów tego artykułu. KG i SB. KG i MM,
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone w przypadku braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
recenzent i redaktor prowadzący zadeklarowali wspólną przynależność.
podziękowania
badania zgłoszone w tej publikacji zostały wsparte przez National Institutes of Health pod numerami nagród CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) i T32 CA009171 (TB)., Za treść odpowiedzialność ponoszą wyłącznie autorzy i niekoniecznie reprezentują oficjalne poglądy National Institutes of Health.
3. Humpton TJ, Vousden KH. Regulacja metabolizmu komórkowego i niedotlenienia przez p53. Cold Spring Harbect Med (2016) 6(7):211-30. podoba mi się! do obserwowanych nr: 101101a026146
CrossRef Full Text | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin m, Liu J, et al., Supresor guza p53 negatywnie reguluje glikolizę stymulowaną przez hipoksję poprzez jej docelowy RRAD. Oncotarget (2014) 5(14):5535-46. podoba mi się! do obserwowanych nr: 186322137
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Lee M, Yoon JH. Metaboliczne wzajemne oddziaływanie glikolizy i utleniania mitochondrialnego: odwrotny efekt Warburga i jego implikacja terapeutyczna. World J Biol Chem (2015) 6(3):148-61. podoba mi się! do obserwowanych nr:104331v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., Wyczerpanie nowego p53-docelowego genu karnityny palmitoiltransferaza 1C opóźnia wzrost guza w modelu guza nerwiakowłókniakowatości typu I. Śmierć Komórki(2013) 20(4):659-68. podoba mi się! do obserwowanych nr:1010382012.168
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari m, Bradley DR, Gleave ME, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo Jl, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in myszy z chimerycznym ludzkim / mysim genem p53 rozwijają się normalnie i wykazują Dzikie odpowiedzi typu P53 na czynniki uszkadzające DNA: nowe biomedyczne narzędzie badawcze. Onkogen (2001) 20(3):320-8. podoba mi się! do obserwowanych nr:101038onc.1204080
PubMed Abstract | CrossRef Full Text | Google Scholar
57. Liu DS, Duong CP, Haupt s, Montgomery KG, House CM, Azar WJ, et al. Hamowanie układu oś xC – / glutationu wybiórczo celuje w nowotwory z akumulacją mutanta-p53., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















