Genome Spot (Polski)
Pathway analysis has become such a common procedure in bioinformatics, especially in studying gene expression. Jeśli spojrzeć na badanie ostatnich gazet wydaje się, że istnieje kilka sposobów, że można to zrobić. W tym poście omówię różnice między dwoma powszechnie używanymi narzędziami; DAVID i GSEA.

pojęcia stojące za tymi dwoma algorytmami są bardzo różne., DAVID określa pokrywanie się między listami genów dostarczonymi przez użytkownika a bazami danych, szukając pokrywania się, które są większe niż oczekiwane przez przypadek. Możesz poprawić dokładność algorytmu, dostarczając plik w tle, który zawiera wszystkie geny, które zostały uznane / wykryte w eksperymencie. Zestawy genów dostarczane przez użytkownika są zwykle generowane przez wybór genów, które przechodzą próg istotności. Procedura DAVID jest podobna do innych dostępnych, takich jak pomysłowość, AmiGO i GeneGO., Wybór wartości istotności jest w dużej mierze arbitralny, ale często ustawia się próg na FDR skorygowany p< 0.05. Zmiana progu znacznie zmieni wyniki wzbogacenia. Stosowane badania statystyczne obejmują hipergeometrię, dokładność Fishera i Chi-kwadrat.
w przeciwieństwie do powyższych metod, które wymagają list genów, które przechodzą dowolne progi, GSEA jest narzędziem wykorzystującym każdy punkt datapoint w swoim algorytmie statystycznym. W” klasycznej ” metodzie geny są uszeregowane według od najbardziej regulowanych w górę do najbardziej regulowanych w dół., Sama metryka rang jest różna, ale dwie ważne metody wykorzystują podpisaną wartość p, lub niższy 90% przedział ufności zmiany krotnie. Podstawą badania jest ocena, czy członkowie zestawu genów wydają się wzbogaceni na jednym końcu profilu. Aby przetestować wzbogacenie, GSEA wykonuje permutacje profilu, obliczając wzbogacenie genu ustawionego tysiąc lub więcej razy, aby empirycznie oszacować wartości P. Inne narzędzia wykorzystujące cały profil wykonują testy analityczne, takie jak test Wilcoxona i Kołmogorowa-Smirnowa.,
więc teraz, gdy rozumiemy główne różnice w algorytmach, możemy zacząć porównywać wyniki z tych dwóch technik. Wróciłem do zbioru danych, o którym mówiłem wcześniej, leczenie azacytydyną w komórkach AML3 profilowanych przez RNA-seq. Użyję różnicowych danych ekspresji RNA-seq wygenerowanych przez DESeq jako punktu wyjścia do porównania Davida i GSEA.
dla Davida zrobiłem plik w tle zawierający wszystkie 19 030 genów wykrytych w eksperymencie powyżej progu wykrywalności. Znacząca (FDR<0.,05) listy genów w kierunku up (2502 genów) i down (2907 genów) zostały oddzielnie przedstawione Davidowi 6.7 do analizy, koncentrując się wyłącznie na szlakach KEGG.
w przypadku GSEA zastosowałem tę samą procedurę metryki Rang opisaną wcześniej. Użyłem klasycznego algorytmu GSEA z gsea2-2.2.2.jar wykonywalny, przy użyciu zestawów genów KEGG z MSigDB v5. 1.
na poniższym wykresie określiłem nakładanie się statystycznie istotnych zestawów genów (FDR<0.05) oddzielnie dla dróg regulowanych w górę i w dół., Uwzględniłem również przecięcie wyników Davida, gdy nie ma tła (tylko dla ciekawości, nie jest to zalecana procedura).
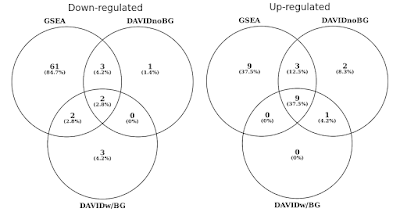
DAVID vs GSEA using the same gene expression profile. Liczby reprezentują zbiory genów, które są znacznie wzbogacone (FDR<0.05).
zauważysz, że zarówno w górę, jak iw dół, GSEA identyfikuje bardziej znaczące wyniki w porównaniu z Davidem. Okazało się, że było to również w przypadku innych bibliotek zestawu genów, takich jak REACTOME I GO., Różnica w czułości między dwoma narzędziami sprowadza się do różnych algorytmów, z DAVID za pomocą dyskretnej listy genów, w porównaniu do GSEA, który wykorzystuje każdy punkt danych. GSEA będzie znacznie bardziej wrażliwe w identyfikacji zestawów genów, które mają wiele członków, które przechodzą subtelne zmiany w ekspresji. Indywidualnie zmiany te nie są istotne statystycznie, ale gdy duża część genów w zbiorze zbiorowo zmienia ekspresję, trend ten może być bardzo istotny statystycznie.,
oprócz bardziej czułego algorytmu, zestawy genów i adnotacje genów Davida są naprawdę nieaktualne. Ostatnia aktualizacja wielu źródeł danych Davida jest w połowie do końca 2009 roku. W związku z tym wiele genów z Ensembl może nie być wspieranych. Rzeczywiście, z mojego pliku w tle, który zawierał 19,030, tylko 13,488 zostało rozpoznanych przez Dawida. Problemem jest również brak zaktualizowanych zestawów genów, podobnie jak stale rosnąca liczba cytowań, które DAVID wciąż otrzymuje.,
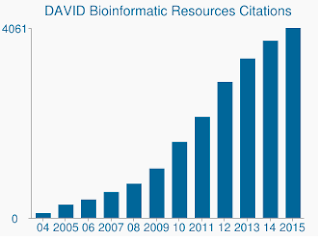
DAVID nadal otrzymuje tysiące cytowań rocznie, mimo że podstawowe dane są żałośnie nieaktualne.
podsumowując, zalecałbym badaczom stosowanie narzędzi, które patrzą na cały profil w celu określenia wzbogacenia (GSEA, CAMERA, WilcoxGST, Pathifier), zamiast przedkładania zestawów genów do analizy nakładania się. Korzystanie z DAVID może być szybką i łatwą opcją, ale odbywa się to kosztem ogromnej redukcji czułości.
Czytaj dalej
JA Timmons, KJ Szkop, IJ Gallagher., Wiele źródeł błędu mylą analizy wzbogacenia funkcjonalnego danych globalnych-omics. Biologia genomu, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Wpływ akumulacji wiedzy na analizę wzbogacania ścieżki. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: w maju 2016 roku DAVID został zaktualizowany!
https://david.ncifcrf.gov/content.jsp?file=release.html















