Genom Spot
Vej analyse er blevet sådan en fælles procedure i bioinformatik, især i at studere genekspression. Hvis man ser på en undersøgelse af de seneste papirer ser det ud til, at der er en masse måder, at det kan gøres. I dette indlæg vil jeg diskutere forskellene mellem to almindeligt anvendte værktøjer; DAVID og GSEA.

begreberne bag de to algoritmer er meget forskellige., DAVID bestemmer overlapninger mellem brugerleverede genlister og de kuraterede databaser, på udkig efter overlapninger, der er større end forventet ved tilfældig chance. Du kan forbedre algoritmens nøjagtighed ved at give en baggrundsfil, der indeholder alle gener, der blev overvejet/detekteret i eksperimentet. De brugerleverede gensæt genereres normalt ved at vælge gener, der passerer en signifikantgrænse. DAVID-proceduren ligner andre tilgængelige som Ingenuity, AmiGO og GeneGO., Udvælgelsen af signifikansværdier er stort set vilkårlig, men det er almindeligt at indstille tærsklen til FDR-justeret p<0.05. Ændring af tærsklen vil ændre berigelsesresultaterne betydeligt. De statistiske tests ansat omfatter hypergeometric, Fisher ‘ s eksakte og Chi-kvadreret.
i modsætning til de ovennævnte metoder, der kræver lister over gener, der passerer vilkårlige tærskler, GSEA er et værktøj, der bruger hver datapoint i sin statistiske algoritme. I den” klassiske ” metode er gener rangeret efter Fra mest opregulerede til mest nedregulerede., Den rang metriske selv varierer, men to gyldige metoder er at bruge underskrevet p-værdi, eller lavere 90% konfidensinterval af folden forandring. Grundlaget for testen er at vurdere, om medlemmer af et gensæt synes beriget i den ene ende af profilen. For at teste berigelsen udfører GSEA permutationer af profilen og beregner berigelsen af genet tusind eller flere gange for at estimere p-værdier empirisk. Andre værktøjer, der bruger hele profilen, udfører analytiske tests som testilco .on og Kolmogorov-Smirnov testen.,
Så nu hvor vi forstår de vigtigste forskelle i algoritmerne, kan vi begynde at sammenligne resultaterne fra disse to teknikker. Jeg gennemgik et datasæt, jeg har diskuteret før, A .acitidinbehandling i AML3-celler profileret af RNA-se.. Jeg vil bruge differential RNA-se.ekspressionsdata genereret af Dese. som et startpunkt for at sammenligne DAVID og GSEA.
for DAVID lavede jeg en baggrundsfil bestående af alle 19.030 gener, der blev påvist i eksperimentet over detektionstærsklen. Den signifikante (FDR<0.,05) genlister i retningen up (2502 gener) og Do .n (2907 gener) blev separat sendt til DAVID 6.7 til analyse, kun med fokus på KEGG-veje.
for GSEA brugte jeg den samme rang metriske procedure beskrevet tidligere. Jeg brugte den klassiske GSEA algoritme med gsea2-2.2.2.jar eksekverbar, ved hjælp af KEGG gen sæt fra MSigDB v5. 1.
i nedenstående graf bestemte jeg overlapningen af statistisk signifikante gensæt (FDR<0.05) separat for op og ned regulerede veje., Jeg inkluderede også et kryds mellem DAVID-resultater, når der ikke gives nogen baggrund (Kun for nysgerrighed, ikke en anbefalet procedure).
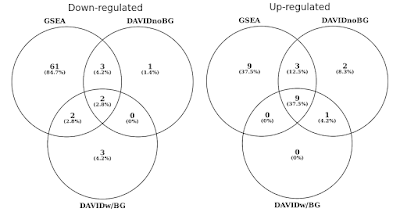
DAVID vs GSEA under anvendelse af den samme genekspressionsprofil. Tal repræsenterer gensæt, der er signifikant beriget (FDR<0.05).
du vil bemærke, at i både op og ned reguleret retning, GSEA identificerer mere signifikante resultater i forhold til DAVID. Jeg fandt, at dette også var tilfældet i andre gensætbiblioteker som REACTOME og GO., Forskellen i følsomheden mellem de to værktøjer koger ned til de forskellige algoritmer, hvor DAVID bruger en diskret liste over gener, versus GSEA, der bruger hvert datapunkt. GSEA vil være langt mere følsomme i at identificere gensæt, der har mange medlemmer, der gennemgår subtile ændringer i udtryk. Individuelt er disse ændringer ikke statistisk signifikante, men når en stor brøkdel af gener i et sæt kollektivt skifter i udtryk, kan denne tendens være meget signifikant statistisk set.,
ud over den mere følsomme algoritme er Davids gensæt og genannotationer virkelig forældede. Den sidste opdatering af Davids mange datakilder er i midten til slutningen af 2009. Som sådan understøttes mange gentiltrædelser fra Ensembl muligvis ikke. Ja, fra min baggrund fil, der indeholdt 19.030 kun 13.488 blev anerkendt af DAVID. Manglen på opdaterede gensæt er også en bekymring, ligesom det stadigt voksende antal citater, som DAVID stadig modtager.,
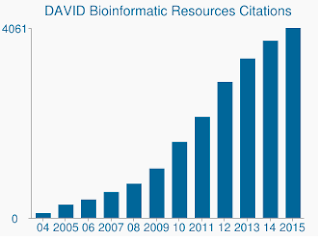
DAVID modtager stadig tusinder af citater om året, på trods af at underliggende data er sørgeligt forældede.
konklusionen er, at jeg vil anbefale forskere bruge værktøjer, der ser på en hel profil til at bestemme berigelse (GSEA, KAMERA, WilcoxGST, Pathifier) hellere end at indsende gen-sæt til overlap analyse. Brug af DAVID kan være den hurtige og nemme mulighed, men dette kommer på bekostning af en massiv reduktion i følsomheden.
yderligere læsning
JA Timmons, KJ s .kop, IJ Gallagher., Flere kilder til bias forvirrer funktionel berigelsesanalyse af globale omics-data. Genombiologi, 2015. 16:186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Videnakkumuleringens indvirkning på vejberigningsanalysen. BIOR .iv. doi: http://dx.doi.org/10.1101/049288
Rediger: i maj 2016 blev DAVID opdateret!
https://david.ncifcrf.gov/content.jsp?file=release.html















