Frontiers en Endocrinología
Introducción
el gen supresor tumoral TP53 ha sido el gen humano más estudiado desde su descubrimiento hace casi 40 años (1). La razón principal detrás de este estado es el papel crítico que desempeña el p53 en la prevención del desarrollo del cáncer, y es ampliamente considerado como el «guardián del genoma».,»Durante algún tiempo se ha creído generalmente que el papel de p53 en la supresión de tumores es en virtud de su capacidad para inducir la apoptosis, la detención del ciclo celular y la senescencia de las células precancerosas (2). Sin embargo, ahora está cada vez más claro que p53 regula muchas otras vías en la célula y que estas otras vías también juegan un papel en la capacidad de p53 para funcionar como un supresor tumoral (3). En particular, el papel de p53 en la regulación de genes implicados en metabolismo y ferroptosis se ha implicado en su capacidad de suprimir el desarrollo del tumor., La Ferroptosis es una nueva vía de muerte celular caracterizada por primera vez en 2012 y puede describirse mejor como una forma de muerte celular dependiente del hierro, independiente de la caspasa, impulsada por la formación de peroxidación lipídica (4). Específicamente, dos modelos de ratón que contienen mutaciones de ingeniería en p53 que eliminan la capacidad de p53 Para inducir la apoptosis y la senescencia conservan la capacidad de suprimir el desarrollo tumoral espontáneo; ambos mutantes conservan la capacidad de transactivar genes en el metabolismo y la ferroptosis (5, 6)., A continuación se detalla un resumen de los datos que implican a p53 en la regulación del metabolismo y la ferroptosis.
Wild-Type (WT) p53 regula positivamente la fosforilación oxidativa y suprime el metabolismo de la glucosa
Wild-type p53 regula la versatilidad metabólica de las células favoreciendo la respiración mitocondrial sobre la glucólisis, en parte a través de la transactivación de SCO2 (citocromo C oxidasa assembly), que desempeña un papel directo en la fosforilación oxidativa (7)., p53 también regula directamente la transactivación de GLS2( Glutaminasa 2); esta enzima permite el uso de glutamina como fuente de energía para las mitocondrias (8). Además, WT p53 regula negativamente la glucólisis reprimiendo transcripcionalmente los transportadores de glucosa GLUT1 y GLUT4, y transactivando RRAD y TIGAR; ambos son inhibidores de la glucólisis (9-11). Finalmente, p53 también se une e inhibe directamente la enzima glucosa-6-fosfato deshidrogenasa, suprimiendo así el metabolismo de la glucosa (12)., De estos y otros estudios se desprende claramente que en organismos normales, no estresados, el p53 regula directamente el estado metabólico en una célula (Figura 1). No es sorprendente que este gen y muchos de sus reguladores estén implicados en enfermedades metabólicas, como la obesidad y la diabetes (13).
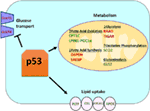
la Figura 1. The role of wild-type (WT) p53 in metabolism. Los Genes regulados positivamente por p53 se muestran en verde, y los genes regulados negativamente por p53 se muestran en rojo., p53 inhibe el transporte de glucosa, la glucólisis y la síntesis de ácidos grasos, mientras que promueve la captación de lípidos, la oxidación de ácidos grasos, la fosforilación oxidativa y la glutaminólisis.
El Mutante p53 regula positivamente el metabolismo de Warburg (glucólisis aeróbica)
en contraste con la función de WT p53, el mutante p53 en las células tumorales favorece la glucólisis aeróbica, en parte aumentando el tráfico del transportador de glucosa GLUT1 a la membrana plasmática, aumentando así la importación de glucosa (14, 15)., Después de la mutación de p53, los niveles reducidos de SCO2 y GLS2 y los niveles aumentados de GLUT1 y GLUT4 favorecen la glucólisis aeróbica sobre la fosforilación oxidativa. De esta manera, se cree que el mutante p53 contribuye a la propensión de las células tumorales a utilizar la glucólisis aeróbica en favor de la fosforilación oxidativa, o el llamado metabolismo Warburg (15). Una de las características distintivas del cáncer es el metabolismo desregulado, generalmente demostrado por este cambio de la glucólisis aeróbica a la fosforilación oxidativa., Aunque esto resulta en un rendimiento de ATP menor y menos eficiente, se cree que las células cancerosas se benefician al desviar los intermedios glicolíticos a las vías biosintéticas necesarias para una rápida división celular (16). Este cambio metabólico también conduce a una disminución de la apoptosis mediada por las mitocondrias y a una señalización más eficiente a través de los metabolitos disponibles en las células cancerosas (17).
una variante genética común en TP53 influye en su función en el metabolismo
hay un polimorfismo común de la región codificante de p53 en el codón 72, codificando para prolina (P72) o arginina (R72)., Esta variación de aminoácidos puede afectar la función p53 con respecto al destino celular después del estrés. En respuesta al daño del ADN, la variante P72 de p53 desencadena predominantemente la detención del ciclo celular, mientras que la variante R72 induce predominantemente la muerte celular, o apoptosis (18, 19). A pesar de estas diferencias en la función, la variación del codón 72 no se ha asociado de manera consistente con la susceptibilidad al cáncer (20). Por el contrario, en estudios en humanos este polimorfismo está significativamente asociado con un aumento del índice de masa corporal y el riesgo de diabetes (21, 22)., Esta premisa está respaldada por estudios en ratones, donde un modelo de ratón para estas variantes del codón 72 muestra un aumento de la diabetes inducida por la dieta alta en grasas en ratones con la variante R72, en comparación con P72. En estos estudios, los genes diana p53 TNFa y NPC1L1 fueron identificados como reguladores críticos en el aumento de la obesidad inducida por la dieta en ratones R72 (23). Curiosamente, también se ha demostrado que la variante R72 confiere una mayor supervivencia de las células en respuesta a la privación de nutrientes (24)., Estos hallazgos han llevado a la hipótesis de que la variante R72 de p53 surgió y se seleccionó a medida que las poblaciones migraban al norte, donde el clima frío requeriría una mayor acumulación de grasa, pero donde la supervivencia en respuesta a la privación de nutrientes también estaría bajo Selección (24).
p53 regula el metabolismo lipídico
aunque p53 es bien conocido por regular la glucólisis y el ciclo del ácido cítrico, p53 también ha demostrado desempeñar un papel en la regulación del metabolismo lipídico (25)., Se cree que WT p53 mejora la oxidación de ácidos grasos mientras inhibe la síntesis de ácidos grasos, actuando así como un regulador negativo de la síntesis de lípidos (25). Hay varios genes de la blanco p53 con papeles en metabolismo del lípido. Sánchez-Macedo y sus colegas demostraron que la carnitina palmitoiltransferasa 1C (CPT1C) está regulada transcripcionalmente por p53; esta enzima ayuda en el transporte de ácidos grasos activados a las mitocondrias., En apoyo de la función de este gen regulado por el p53 en el cáncer, este grupo mostró que los ratones con deficiencia de Cpt1c muestran un retraso en el desarrollo tumoral y tasas de supervivencia más altas (26). La Lipina 1 (LPIN1) es otro gen diana de p53; la LPIN1 es necesaria para el desarrollo adecuado de adipocitos y se induce en condiciones de bajos nutrientes (27). Finck y sus colegas mostraron que la LPIN1 interactúa con PGC-1α, otro gen blanco conocido de p53 con un papel en el metabolismo, y que esta interacción activa la expresión de genes involucrados en la promoción de la oxidación de ácidos grasos (28).,
además de regular directamente la transcripción de genes involucrados en el metabolismo lipídico, p53 también puede regular el metabolismo lipídico de una manera que implica la interacción directa proteína–proteína. Por ejemplo, la glucosa-6-fosfato deshidrogenasa, que es la enzima limitante de la velocidad en la vía de la pentosa fosfato, se une y es inhibida directamente por p53, lo que resulta en una disminución de la producción de NADPH y, en consecuencia, en una disminución de la síntesis de ácidos grasos (12)., La familia de factores de transcripción SREBP (sterol regulatory element-binding proteins) modula la expresión de genes involucrados en la síntesis de colesterol, ácidos grasos, triacilglicerol y fosfolípidos (29-31). WT p53 reprime la función de SREBP (32), mientras que las formas mutantes de p53 se unen directamente a SREBP y mejoran su función transcripcional, lo que lleva a un aumento de la actividad de SREBP en tumores humanos (33, 34). En consecuencia, el mutante p53 se correlaciona con una mayor expresión de genes de biosíntesis de esteroles en tumores de mama humanos (34, 35)., Finalmente, la proteína quinasa activada por AMP (AMPK) es una enzima que se activa bajo niveles bajos de nutrientes o estrés energético y se sabe que inhibe la síntesis de ácidos grasos al interactuar con la acetil-CoA-carboxilasa y SREBP-1 (36, 37). Zhou y sus colegas demostraron que el mutante p53 se une preferentemente e inhibe la AMPK, lo que lleva a un aumento de la síntesis de ácidos grasos. Como resultado, las proteínas p53 mutantes conducen a un aumento de la señalización AMPK, contribuyendo al crecimiento celular invasivo de las células tumorales (33). Un área menos explorada es el papel de p53 en el transporte de lípidos., Se ha demostrado que p53 regula transcripcionalmente la apolipoproteína B (apoB) y el complejo enzimático de edición apoB 1, lo que indica el papel de p53 en la regulación de las lipoproteínas aterogénicas (38). El análisis de microarrays de células humanas derivadas del hígado identificó la proteína de transferencia de fosfolípidos, el casete de unión a ATP A12 y la Carboxil éster lipasa como tres genes diana p53 que desempeñan un papel en el transporte de lípidos (39, 40)., En general, aunque está claro que el p53 juega un papel clave en la mediación de la síntesis de lípidos y el metabolismo, la contribución de esta vía, y estos genes diana de p53, a la supresión tumoral por p53 aún está por determinar (Figura 1).
La Ferroptosis es una nueva vía de muerte celular impulsada por la peroxidación lipídica
en 2012, Dixon y sus colegas descubrieron una nueva forma de muerte celular regulada llamada ferroptosis. La Ferroptosis es una forma de muerte celular dependiente del hierro, independiente de la caspasa, resultante de la acumulación de lípidos oxidados (4, 41)., Este proceso es impulsado por la inactivación de la glutatión peroxidasa 4 (GPX4), una enzima responsable de convertir los hidroperóxidos lipídicos letales en alcoholes lipídicos no tóxicos, que requiere glutatión para funcionar (41). Se cree que la peroxidación de los ácidos grasos poliinsaturados (AGPI) es el impulso impulsor de la muerte celular por ferroptosis. Los AGPI contienen protones bis-alílicos que pueden ser fácilmente abstraídos y producen radicales que reaccionarán con el oxígeno, creando más Radicales y dando como resultado una reacción en cadena de especies reactivas de oxígeno lipídico (42)., El mecanismo exacto de la muerte celular por ferroptosis sigue siendo desconocido, pero una hipótesis es que el daño lipídico conduce a la destrucción de la membrana plasmática (43). Se ha especulado que la ferroptosis podría ser un mecanismo de supresión de tumores que funciona mediante la eliminación de células que están privadas de nutrientes o que han estado expuestas a un estrés ambiental o infección.,
regulación farmacológica de la Ferroptosis
La Ferroptosis se puede inducir utilizando inhibidores del sistema xc, como erastin, o análogos como glutamato y sorafenib, que inhiben la importación de cistina, lo que resulta en glutatión agotado y la posterior inactivación de GPX4. Alternativamente, la ferroptosis puede ser inducida por (1S,3R)-RSL3 (en lo sucesivo denominado RSL3), que se une directamente e inhibe GPX4 (4, 5, 42). Buthione sulfoximine, FIN56, FINO2, CCl4, y cisplatin son otros agentes que se han demostrado para inducir ferroptosis en células., La muerte por ferroptosis se puede prevenir mediante la supresión de la peroxidación lipídica, que se puede lograr mediante el uso de antioxidantes lipofílicos, como ferrostatina-1, liproxstatina-1, o vitamina E. quelantes de hierro como deferoxamina o cicloprox son otra herramienta utilizada para suprimir la ferroptosis mediante la reducción de los niveles de hierro. El agotamiento de los AGPI o la adición de ácidos grasos monoinsaturados a los medios de cultivo celular también pueden rescatar a las células de la ferroptosis (42, 44).,
La Ferroptosis está implicada en la supresión tumoral mediada por p53
en 2012, Gu y sus colegas desarrollaron un modelo de ratón en el que tres residuos de lisina normalmente acetilados en el dominio de unión al ADN de p53 fueron mutados a arginina, y por lo tanto no pudieron ser acetilados; este ratón se conoce como el ratón 3KR. Notablemente, las células del ratón 3KR no pueden experimentar apoptosis dependiente de p53, detención del ciclo celular o senescencia, y de hecho el mutante 3KR de p53 no logra transactivar la mayoría de los genes diana de p53., Curiosamente, este modelo de ratón no desarrolla cáncer espontáneamente, lo que implica que p53 podría suprimir el desarrollo tumoral independientemente de la senescencia o la apoptosis (45). Este grupo encontró que la proteína mutante 3KR retiene la capacidad de someterse a ferroptosis y regular el metabolismo de la cistina mediante la regulación de la expresión del importador de cistina SLC7A11; esto sugirió que la ferroptosis podría ser una vía que subyace a la supresión tumoral mediada por p53., Cuando los MEF de tipo salvaje y 3kr fueron tratados con el inductor de ferroptosis Erastin, se observó casi un 50% de muerte celular, mientras que los MEF nulos de p53 exhibieron un 20% de muerte celular; esto indica que p53 sensibiliza a las células a la ferroptosis, y también que otros reguladores clave también juegan un papel en la ferroptosis (5). Posteriormente, gu y sus colegas identificaron un sitio de acetilación adicional en lisina 98 de p53, y generaron un modelo de ratón en el que los cuatro sitios de acetilación fueron mutados a arginina (4KR)., Curiosamente, el mutante 4KR fue incapaz de regular los genes involucrados en la ferroptosis como SLC7A11, y a diferencia del mutante 3KR fue incapaz de suprimir el desarrollo tumoral (46). Aunque en la actualidad son correlativos, estos datos implican el papel de p53 en la ferroptosis en su capacidad para suprimir el desarrollo tumoral.
en células no transformadas, p53 regula positivamente la Ferroptosis
Además de SLC7A11, se ha descubierto que varios otros genes diana directos p53 juegan un papel en la ferroptosis. Estos incluyen GLS2, PTGS2 y SAT1., Los estudios de dos grupos separados apoyan el papel de GLS2 en la ferroptosis, que se sabe que disminuye el glutatión y aumenta los niveles celulares de ROS. Jiang y sus colegas usaron inhibidores de ferroptosis combinados con inhibidores de glutaminólisis para inhibir la ferroptosis inducida por Erastina, demostrando así que la ferroptosis requiere glutaminólisis y GLS2 (47). Murphy y sus colegas mostraron que una variante polimórfica de p53 fue capaz de inducir la detención del crecimiento y la senescencia en las células humanas y murinas, pero no pudo reprimir SLC7A11 o transactivar GLS2., Esta variante tuvo un marcado deterioro en la inducción de la ferroptosis y la supresión del desarrollo tumoral, lo que nuevamente implicó el papel de p53 en la supresión tumoral mediada por ferroptosis (48). Otro gen objetivo p53 con un papel en la ferroptosis es PTGS2, un gen que codifica la enzima ciclooxigenasa-2. Stockwell y colegas primero mostraron que la inducción de ferroptosis usando Erastin y RSL3 condujo a la regulación ascendente de PTGS2 (41). Notably, PTGS2 was not upregulated by ferroptosis inducers in p53-null cells, suggesting that this regulation is p53 dependent (5)., Actualmente, la regulación ascendente de PTGS2 es ampliamente utilizada como marcador de ferroptosis (5, 41).
un estudio reciente del grupo Gu mostró que el gen diana p53 SAT1 regula la ferroptosis (49). Los autores identificaron SAT1 como Blanco directo de p53 y mostraron que el silenciamiento de SAT1 redujo la muerte celular inducida por especies reactivas de oxígeno en células con peso p53, pero no tuvo efecto en células p53-nulas. Mecánicamente, este grupo mostró que SAT1 aumenta el nivel y la actividad de la araquidonato 15-lipoxigenasa, una enzima de unión al hierro que oxida los AGPI y aumenta la peroxidación lipídica., Notablemente, este estudio mostró que ni p53 ni SAT1 por sí solos parecen ser suficientes para inducir ferroptosis. En cambio, los datos combinados son más consistentes con la premisa de que p53, en virtud de la regulación de los genes que contribuyen a la ferroptosis, regula la sensibilidad de las células a esta vía, en lugar de inducir directamente la ferroptosis. Queda por determinar si el p53 regula otros genes implicados en la ferroptosis (Figura 2).
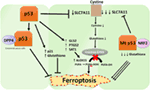
la Figura 2. The various roles of p53 in ferroptosis., La inhibición de la glutatión peroxidasa 4 (GPX4), la enzima clave que cataliza la conversión de ácidos grasos poliinsaturados (AGPI) que contienen peróxidos a alcoholes, es el principal impulsor de la ferroptosis. Dependiendo del contexto, p53 puede suprimir la ferroptosis (como en las células cancerosas colorrectales) o promover la ferroptosis. El mutante p53 sensibiliza a las células a la ferroptosis incluso más que el tipo salvaje p53.,
en algunas células, p53 regula negativamente la Ferroptosis
Un estudio recientemente publicado por Tarangelo y sus colegas muestra que p53 regula negativamente la ferroptosis en células cancerosas (50). Este grupo encontró que el pre-tratamiento de células con Nutlin-3, un compuesto que estabiliza p53 retrasa el inicio de la ferroptosis en varios tipos de células. Se encontró que el inicio retardado de la ferroptosis dependía de CDKN1A (codificación p21), un objetivo crítico de transcripción p53., El mecanismo a través del cual P21 retrasa la ferroptosis aún no se ha dilucidado, pero se cree que la conservación del glutatión intracelular puede ser un factor que contribuye a la reducción de la sensibilidad a la ferroptosis. Los autores concluyen que el eje p53-p21 permite a las células cancerosas sobrevivir en condiciones de estrés metabólico, como la privación de cistina, al suprimir la aparición de ferroptosis (50). Un estudio reciente mostró que p53 inhibe la ferroptosis en las células de cáncer colorrectal mediante la unión a la enzima dipeptidil-peptidasa-4 (DPP4), que es un modulador de la ferroptosis y el metabolismo de los lípidos., Mecánicamente, este estudio mostró que p53 antagoniza la ferroptosis al secuestrar DPP4 en un grupo inactivo enzimático nuclear. En ausencia de p53, DPP4 es libre de interactuar y formar un complejo con NOX1; esto conduce a un aumento de la peroxidación lipídica y ferroptosis. La inhibición de DPP4 suprime la ferroptosis de manera significativa, mientras que la sobreexpresión de DPP4 desencadena la sensibilidad a la Erastina, particularmente en células agotadas por p53 (51). El control bidireccional de la ferroptosis por p53 a través de mecanismos dependientes de la transcripción e independientes de la transcripción puede ser dependiente del contexto o del tipo celular (Figura 2).,
el polimorfismo P47S de TP53 afecta la Ferroptosis y la supresión tumoral
Además de las mutaciones sin sentido, hay varios polimorfismos de nucleótido único (SNPs) funcionalmente significativos en el gen TP53 y otras proteínas conocidas por regular esta vía (como MDM2 y MDM4). La variante Pro47Ser (en adelante S47) es el segundo SNP más común encontrado en la región de codificación p53 (después de Pro72Arg) que altera la secuencia de aminoácidos de la proteína., Para dilucidar mejor el impacto de esta variante en la función p53 y el riesgo de cáncer, El Grupo Murphy generó un modelo de ratón p53 humanizado, en el que los exones 4-9 del p53 Murino fueron reemplazados por exones p53 humanos que contenían el tipo salvaje o la variante S47 (52-55). La mayoría de los ratones S47 desarrollaron espontáneamente tumores de varios tipos histológicos, particularmente cáncer de hígado, entre los 12 y 18 meses de edad, a diferencia de los ratones WT p53 (48)., En fibroblastos embrionarios de ratón y líneas celulares linfoblastoides humanas, la variante S47 mostró deterioro de la muerte celular programada en respuesta al cisplatino y otras tensiones genotóxicas. Mecánicamente, la variante S47 es defectuosa para la transactivación de genes involucrados en el metabolismo, como Gls2 (glutaminasa 2) y Sco2 (48). De acuerdo con el papel de Gls2 en la ferroptosis, este grupo encontró que las células S47 eran marcadamente resistentes a los agentes inductores de ferroptosis Erastin y RSL3 (47, 48). Este defecto puede contribuir al fenotipo propenso al tumor observado en ratones S47.,
El Mutante p53 sensibiliza las células tumorales a la Ferroptosis
El tipo salvaje p53 regula negativamente la expresión del importador de cistina SLC7A11, que inhibe la sensibilidad a la ferroptosis (5). Aunque esta regulación ocurre en las células normales, en las células tumorales, otros mediadores de SLC7A11 parecen predominar en la regulación de este gen. Por ejemplo, el factor de transcripción antioxidante maestro NRF2 también puede regular la expresión de SLC7A11 a nivel transcripcional, y NRF2 ha sido implicado como un jugador clave en la protección de las células cancerosas contra la ferroptosis., Por ejemplo, la inhibición de NRF2 en células cancerosas hepatocelulares aumenta la actividad anticancerosa de Erastin y Sorafenib in vivo (56). Las formas mutantes de p53 pueden inhibir la función NRF2 por interacción directa, y un grupo encontró que los tumores con p53 mutante contienen niveles muy bajos de SLC7A11, y por lo tanto muestran una mayor sensibilidad a la ferroptosis. En particular, la sobreexpresión de SLC7A11 en modelos de p53 mutante condujo a la resistencia a los medicamentos, lo que sugiere que los niveles de expresión de SLC7A11 deben considerarse cuando se dirigen a cánceres impulsados por p53 mutante con compuestos inductores de ferroptosis (57)., En apoyo de esta premisa, un trabajo reciente en cáncer colorrectal (CCR), donde la mutación o deleción de p53 es un evento frecuente, mostró que las líneas celulares de CCR humanas que albergan el mutante p53 eran mucho más sensibles a la muerte celular mediada por Erastin en comparación con las células de CCR con p53 en peso. Para validar estos hallazgos, mostraron que el golpe de una mutación p53 hotspot en las células HCT116 y SW48 restauró la sensibilidad a Erastin (51). Estos datos destacan un nuevo mecanismo por el cual los cánceres impulsados por el mutante p53 pueden ser explotados usando terapia dirigida.,
conclusión
el papel de p53 en el metabolismo es bastante claro y posiblemente incluso intuitivamente obvio: WT p53 limita el metabolismo de la glucosa y la síntesis de lípidos, mientras que mutante p53 parece hacer lo contrario. La contribución de su función metabólica a la supresión tumoral por p53, y a la capacidad del mutante p53 para impulsar la progresión tumoral, aún no se ha demostrado de manera inequívoca. El papel de p53 en la regulación de la ferroptosis, y la contribución de esta función, a la supresión del tumor es aún menos claro., Si bien los datos convincentes de los modelos de ratón apoyan la premisa de que p53 regula la sensibilidad de las células a la ferroptosis, esto puede restringirse a la capacidad de p53 basal para suprimir el desarrollo tumoral espontáneo, y en los modelos de ratón estresados por oncogenes, está claro que la senescencia y la apoptosis juegan el papel predominante. Del mismo modo, p53 puede regular la sensibilidad a la ferroptosis de una manera específica del tipo celular. Se necesitan más estudios en modelos animales, con atención a la ferroptosis en diferentes tejidos, para comprender más plenamente el papel de p53 en la ferroptosis y la ferroptosis en la supresión tumoral., Además, es necesario lograr una idea más clara de qué genes diana p53 desempeñan un papel en la sensibilidad a la ferroptosis. La resolución de estas preguntas debe proporcionar nuevas vías muy necesarias para combatir los tumores con mutante p53.
contribuciones de los autores
KG, SB, TB, AB-K, C-PK y MM escribieron uno o dos párrafos de este artículo. KG y SB hicieron la cifra. KG y MM esbozaron el capítulo.,
Declaración de conflicto de intereses
los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran ser interpretadas como un potencial conflicto de intereses.
El revisor OAF y el editor de manejo declararon su afiliación compartida.
reconocimientos
La investigación reportada en esta publicación fue apoyada por los Institutos Nacionales de salud con los números de premio CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) y T32 CA009171 (TB)., El contenido es responsabilidad exclusiva de los autores y no representa necesariamente las opiniones oficiales de los Institutos Nacionales de la salud.
3. Humpton TJ, Vousden KH. Regulación del metabolismo celular y la hipoxia por p53. Cold Spring Harb Perspect Med (2016) 6(7):211-30. doi: 10.1101 / cshperspect.a026146
CrossRef Texto Completo | Google Scholar
10. Zhang C, Liu J, Wu R, Liang y, Lin M, Liu J, et al., El supresor tumoral p53 regula negativamente la glucólisis estimulada por la hipoxia a través de su RRAD objetivo. Oncotarget (2014) 5(14):5535-46. doi: 10.18632 / oncotarget.2137
PubMed Abstract | CrossRef Texto Completo | Google Scholar
17. Lee M, Yoon JH. Metabolic interplay between glycolysis and mitochondrial oxidation: the reverse Warburg effect and its therapeutic implication. World J Biol Chem (2015) 6(3):148-61. doi: 10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., El agotamiento del nuevo gen diana p53 carnitina palmitoiltransferasa 1C retrasa el crecimiento tumoral en el modelo tumoral de neurofibromatosis tipo I. La Muerte Celular Difiere (2013) 20(4):659-68. doi: 10.1038 / cdd.2012.168
PubMed Abstract | CrossRef Texto Completo | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Desregulación de las proteínas de unión al elemento de respuesta esterólica y efectores posteriores en el cáncer de próstata durante la progresión a la independencia androgénica., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: a new biomedical research tool. Oncogén (2001) 20(3):320-8. doi: 10.1038 / sj.onc.1204080
PubMed Abstract | CrossRef Texto Completo | Google Scholar
57. Liu DS, Duong CP, Haupt s, Montgomery KG, House CM, Azar WJ, et al. Inhibiendo el sistema XC – / glutatión eje selectivamente se dirige a los cánceres con acumulación mutante-p53., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















