Genome Spot (Español)
El análisis de vías se ha convertido en un procedimiento común en Bioinformática, especialmente en el estudio de la expresión génica. Si nos fijamos en una encuesta de artículos recientes parece que hay un montón de maneras que se puede hacer. En este post, voy a discutir las diferencias entre dos herramientas de uso común; DAVID y GSEA.

Los conceptos detrás de los dos algoritmos son muy diferentes., DAVID determina las superposiciones entre las listas de genes proporcionadas por el Usuario y las bases de datos curadas, buscando superposiciones que sean más grandes de lo esperado por el azar. Puede mejorar la precisión del algoritmo proporcionando un archivo de fondo que contenga todos los genes que se consideraron/detectaron en el experimento. Los conjuntos de genes suministrados por el usuario normalmente se generan seleccionando genes que pasan un umbral de significación. El procedimiento DAVID es similar a otros disponibles como Ingenuity, AmiGO y GeneGO., La selección de los valores de significación es en gran medida arbitraria, pero es común establecer el umbral en FDR AJUSTADO p<0.05. La modificación del umbral alterará considerablemente los resultados del enriquecimiento. Las pruebas estadísticas empleadas incluyen hipergeométrica, exacta de Fisher y Chi-cuadrado.en contraste con los métodos anteriores que requieren listas de genes que pasan umbrales arbitrarios, GSEA es una herramienta que utiliza cada punto de datos en su algoritmo estadístico. En el método «clásico» los genes se clasifican de la más regulada hacia arriba a la más regulada hacia abajo., La métrica de rango en sí varía, pero dos métodos válidos son usar el valor p firmado, o un intervalo de confianza del 90% inferior del cambio de pliegue. La base de la prueba es evaluar si los miembros de un conjunto de genes parecen enriquecidos en un extremo del perfil. Para probar el enriquecimiento, GSEA realiza permutaciones del perfil, calculando el enriquecimiento del conjunto de genes mil o más veces para estimar empíricamente los valores de P. Otras herramientas que utilizan el perfil completo realizan pruebas analíticas como la prueba de Wilcoxon y Kolmogorov-Smirnov.,
así que ahora que entendemos las principales diferencias en los algoritmos, podemos comenzar a comparar los resultados de estas dos técnicas. Revisé un conjunto de datos que he discutido antes, el tratamiento con azacitidina en células AML3 perfiladas por ARN-seq. Utilizaré los datos de expresión diferencial de ARN-seq generados por DESeq como punto de partida para comparar DAVID y GSEA.
Para DAVID, hice un archivo de fondo que consta de todos los 19.030 genes detectados en el experimento por encima del umbral de detección. El significativo (FDR< 0.,05) las listas de genes en la dirección up (2502 genes) y down (2907 genes) se presentaron por separado a DAVID 6.7 para su análisis, centrándose solo en las vías KEGG.
para GSEA, utilicé el mismo procedimiento métrico de rango descrito anteriormente. Utilicé el algoritmo gsea clásico con el gsea2-2.2.2.ejecutable jar, usando conjuntos de genes KEGG de MSigDB v5.1.
En el gráfico a continuación, determiné la superposición de conjuntos de genes estadísticamente significativos (FDR<0.05) por separado para vías reguladas hacia arriba y hacia abajo., También incluí una intersección de los resultados de DAVID cuando no se proporciona ningún fondo (solo por curiosidad, no es un procedimiento recomendado).
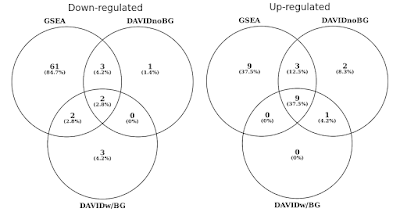
DAVID vs GSEA usando el mismo perfil de expresión génica. Los números representan conjuntos de genes que están significativamente enriquecidos (FDR<0.05).notarás que tanto en la dirección regulada hacia arriba como hacia abajo, GSEA identifica resultados más significativos en comparación con DAVID. Descubrí que este también era el caso en otras bibliotecas de conjuntos de genes como REACTOME y GO., La diferencia en la sensibilidad entre las dos herramientas se reduce a los diferentes algoritmos, con DAVID utilizando una lista discreta de genes, frente a GSEA que utiliza cada punto de datos. GSEA será mucho más sensible en la identificación de conjuntos de genes que tienen muchos miembros que están experimentando cambios sutiles en la expresión. Individualmente, estos cambios no son estadísticamente significativos, pero cuando una gran fracción de genes en un conjunto cambia colectivamente en la expresión, esa tendencia podría ser muy significativa estadísticamente hablando.,además del algoritmo más sensible, los conjuntos de genes y las anotaciones de genes de DAVID están realmente desactualizados. La última actualización de las muchas fuentes de datos de DAVID fue a mediados y finales de 2009. Como tal, muchas accesiones de genes de Ensembl pueden no ser soportadas. De hecho, de mi archivo de antecedentes que contenía 19,030 solo 13,488 fueron reconocidos por DAVID. La falta de conjuntos de genes actualizados también es una preocupación, al igual que el número cada vez mayor de citas que DAVID todavía recibe.,
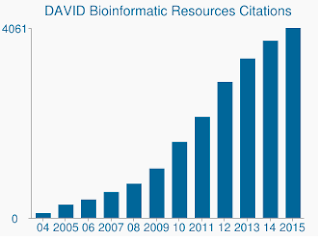
DAVID todavía recibe miles de citas por año a pesar de que los datos subyacentes están lamentablemente desactualizados.
En conclusión, recomendaría a los investigadores utilizar herramientas que miran un perfil completo para determinar el enriquecimiento (GSEA, CAMERA, WilcoxGST, Pathifier) en lugar de enviar conjuntos de genes para el análisis de superposición. El uso de DAVID podría ser la opción rápida y fácil, pero esto viene a expensas de una reducción masiva en la sensibilidad.JA Timmons, KJ Szkop, IJ Gallagher., Múltiples fuentes de sesgo confunden el análisis de enriquecimiento funcional de los datos globales -icsicos. Genome biology, 2015. 16: 186 Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Impact of knowledge accumulation on pathway enrichment analysis. bioRxiv. doi: http://dx.doi.org/10.1101/049288
Editar: en mayo de 2016, DAVID fue actualizado!
https://david.ncifcrf.gov/content.jsp?file=release.html















