Genomin Paikalla
Polulla-analyysi on tullut tällainen yhteinen menettely, bioinformatiikan, erityisesti opiskelu-geenin ilmentyminen. Jos katsoo tuoreiden lehtien kyselyä, niin näyttää siltä, että siinä on monta tapaa, millä se voidaan tehdä. Tässä viestissä aion keskustella kahden yleisesti käytetyn työkalun, Davidin ja GSEAN, eroista.

käsitteiden takana kaksi algoritmit ovat hyvin erilaisia., DAVID määrittää päällekkäisyyksiä käyttäjän antamien geeni luettelot ja kuratoinut tietokantoja, etsiä päällekkäisyyksiä, jotka ovat suurempia kuin odotettavissa sattumaa. Voit parantaa tarkkuutta algoritmi tarjoamalla tausta-tiedosto, joka sisältää kaikki geenit, joita pidettiin/havaittu kokeilu. Käyttäjän toimittamat geenisarjat syntyvät yleensä valitsemalla merkkikynnyksen ylittäviä geenejä. DAVID menettely on samanlainen kuin muut saatavilla, kuten nerokkuus, AmiGO ja GeneGO., Valinta merkitys arvot on pitkälti mielivaltainen, mutta se on tervettä asettaa raja-arvo, FDR oikaistu p<0.05. Kynnyksen muuttaminen muuttaa väkevöintituloksia huomattavasti. Tilastollisiin testeihin kuuluvat hypergeometrinen, Fisherin tarkka ja Chi-neliöinen.
toisin kuin edellä mainitut menetelmät, jotka vaativat luettelot geenejä, jotka kulkevat mielivaltaisen kynnysarvot, GSEA on työkalu, joka käyttää jokaisen datapoint sen tilastollinen algoritmi. ”Klassisessa” menetelmässä geenit asetetaan paremmuusjärjestykseen säädellyimmästä säädellyimmäksi., Rank metriikka itsessään vaihtelee, mutta kaksi kelvollista menetelmää on käyttää allekirjoitettua p-arvoa eli pienempää 90%: n luottamusväliä kertamuutoksesta. Testin lähtökohtana on arvioida, näyttävätkö geenijoukon jäsenet rikastuneina Profiilin toisessa päässä. Testata rikastamiseen, GSEA suorittaa permutaatiot profiilin, laskettaessa rikastuminen geeni asettaa tuhat kertaa tai useammin arvioida p-arvoja empiirisesti. Muut työkalut, jotka käyttävät koko profiilia suorittaa analyyttisiä testejä, kuten Wilcoxon ja Kolmogorov-Smirnov testi.,
joten nyt kun ymmärrämme algoritmien tärkeimmät erot, voimme alkaa vertailla näiden kahden tekniikan tuloksia. Olen uudelleen datajoukon olen keskustellut ennen, atsasitidiini hoito AML3 solujen profiloitu RNA-seq. Käytän DESeq: n tuottamaa differentiaalista RNA-seq-ekspressiotietoa lähtökohtana Davidin ja GSEAN vertailussa.
DAVID, tein taustan-tiedosto joka koostuu kaikista 19,030 geenit havaita kokeessa edellä tunnistus kynnysarvon. Merkittävä (FDR<0.,05) geeni luettelot up (2502 geenit) ja alas (2907 geenit) suuntaan olivat erikseen toimitettu DAVID 6.7 analyysi, jossa keskitytään KEGG väyliä vain.
gsean kohdalla käytin aiemmin kuvattua samaa rank-metristä menettelyä. Käytin klassista gsean algoritmia gsea2-2.2.2: lla.jar executable, käyttäen KEGG geeni asetetaan MSigDB v5.1.
kaaviossa alle, olen päättänyt, päällekkäisyys tilastollisesti merkitsevä geeni sarjaa (FDR<0.05) erikseen ylös-ja alas säännelty väyliä., Otin mukaan myös Daavidin tulosten risteyksen, kun taustoja ei ole (vain uteliaisuudesta, ei suositeltavasta menettelystä).
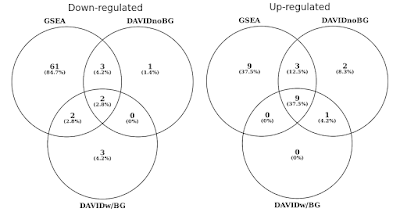
DAVID vs GSEA käyttää saman geenin ilmentyminen profiili. Numerot edustavat merkittävästi rikastuneita geenisarjoja (FDR<0,05).
huomaat, että sekä ylös ja alas säännelty suuntaan, GSEA tunnistaa enemmän merkittäviä tuloksia verrattuna DAVID. Näin oli myös muissa geenisarjakirjastoissa, kuten REACTOMESSA ja GO: ssa., Ero herkkyys välillä kaksi työkaluja kuihtuu eri algoritmeja, DAVID käyttäen diskreetti luettelo geenit, vs. GSEA, joka käyttää jokainen datapiste. GSEA on paljon herkempi tunnistamaan geenin sarjaa, joka on paljon jäseniä, jotka ovat käynnissä hienovaraisia muutoksia ilme. Yksilöllisesti, nämä muutokset eivät ole tilastollisesti merkittäviä, mutta kun suuri osa geenien joukko kollektiivisesti muutos ilmaisu, että trendi voisi olla erittäin merkittävä tilastollisesti.,
lisäksi herkempiä algoritmi, DAVID geenin sarjaa ja gene merkinnät ovat todella vanhentuneita. Viimeisin päivitys Davidin monista tietolähteistä on vuoden 2009 puolivälistä loppuvuoteen. Näin ollen monia geenimuunnoksia ei välttämättä tueta. Todellakin, minun tausta tiedosto, joka sisälsi 19,030 vain 13,488 olivat tunnustettu by DAVID. Myös päivitettyjen geenisarjojen puute huolettaa, samoin yhä kasvava määrä lainauksia, joita DAVID vielä saa.,
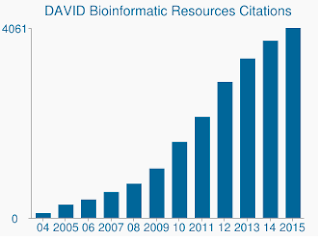
DAVID edelleen vastaanottaa tuhansia lainaukset vuodessa, vaikka taustalla olevat tiedot on valitettavan vanhentunut.
lopuksi, suosittelen tutkijat käyttävät työkaluja, jotka katso koko profiili määrittää rikastamiseen (GSEA, KAMERA, WilcoxGST, Pathifier) mieluummin kuin antaa geenin sarjaa päällekkäin analyysi. David voi olla nopea ja helppo vaihtoehto, mutta tämä tulee kustannuksella massiivinen vähentäminen herkkyys.
Further reading
JA Timmons, KJ Szkop, IJ Gallagher., Useat bias-lähteet sekoittavat toiminnallisen rikastusanalyysin maailmanlaajuisista omics-tiedoista. Genomibiologia, 2015. 16:186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Tietämyksen kertymisen vaikutus reitin rikastusanalyysiin. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: toukokuussa 2016 David päivitettiin!
https://david.ncifcrf.gov/content.jsp?file=release.html















