Rajoja ja Endokrinologian
Johdanto
kasvain vaimennin geeni TP53 on ollut eniten tutkittu ihmisen geeni, koska sen löytö lähes 40 vuotta sitten (1). Tärkein syy statukseen on p53: n keskeinen rooli syövän kehittymisen ehkäisemisessä, ja sitä pidetään yleisesti ”genomin suojelijana.,”Jo jonkin aikaa on yleisesti uskottu, että p53: n rooli kasvaimen tukahduttamisessa johtuu sen kyvystä indusoida apoptoosi, solusyklin pysähtyminen ja esisyöpäsolujen vanheneminen (2). Kuitenkin, se on nyt yhä selvemmäksi, että p53 säätelee monia muita väyliä solussa ja että nämä muut väylät myös pelata rooli p53: n kyky toimia yhtenä kasvain vaimennin (3). Erityisesti p53: n rooli aineenvaihduntaan ja ferroptoosiin osallistuvien geenien säätelyssä on yhdistetty sen kykyyn tukahduttaa kasvaimen kehitystä., Ferroptosis on uusi solukuoleman reitin ensimmäinen tunnettu vuonna 2012, ja se voidaan parhaiten kuvata rauta-riippuvainen, caspase-itsenäinen solun kuolema ohjaa muodostumista lipidiperoksidaation (4). Erityisesti kaksi hiirtä mallit sisältävät suunniteltu mutaatioita p53, joka poistaa kyky p53 aiheuttavan apoptoosia ja vanheneminen sekä säilyttää kyky tukahduttaa spontaani kasvaimen kehitystä; nämä molemmat muunnokset säilyttävät kyvyn transactivate geenien aineenvaihduntaa ja ferroptosis (5, 6)., Seuraavassa on yhteenveto tiedoista, jotka liittyvät p53: een metabolian ja ferroptoosin säätelyssä.
villityypin (WT) p53 Säätelee Positiivisesti Oksidatiivisen Fosforylaation ja Estää Glukoosin Aineenvaihduntaa
Villi-tyyppi p53 säätelee aineenvaihdunnan monipuolisuus solujen suosimalla mitokondrioiden hengitystä yli glykolyysin, osittain kautta transactivation ja SCO2 (sytokromi c oksidaasi assembly), joka on suora rooli oksidatiivisen fosforylaation (7)., p53 säätelee myös suoraan GLS2: n (Glutaminaasi 2) transaktivaatiota; tämä entsyymi mahdollistaa glutamiinin käytön mitokondrioiden energialähteenä (8). Lisäksi WT p53 säätelee negatiivisesti glykolyysin, jonka transcriptionally tukahduttaa glukoosin siirtäjät GLUT1 ja GLUT4, ja transactivating RRAD ja TIGAR; molemmat ovat glykolyysin estäjät (9-11). Lopuksi p53 myös sitoutuu suoraan glukoosi-6-fosfaattidehydrogenaasientsyymiin ja estää sitä, mikä vaimentaa glukoosiaineenvaihduntaa (12)., Se on selvää, nämä ja muut tutkimukset, että normaali, painottomat organismeja, p53 suoraan säätelee aineenvaihdunnan solussa (Kuva 1). Ei ole yllättävää, tämä geeni ja monet sen sääntelyviranomaiset ovat mukana aineenvaihdunnan sairauksiin, kuten lihavuuden ja diabeteksen (13).
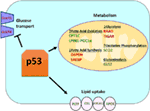
Kuva 1. Villin tyypin (WT) p53: n rooli aineenvaihdunnassa. P53: n positiivisesti säätelemät geenit näkyvät vihreinä, ja p53: n negatiivisesti säätelemät geenit näkyvät punaisina., p53 estää glukoosin kuljetus, glykolyysin ja rasvahappojen synteesi, kun se edistää rasva-kertymä, rasvahappojen hapettumista, oksidatiivinen fosforylaatio, ja glutaminolysis.
Mutant p53 Säätelee Positiivisesti Warburg Aineenvaihduntaa (Aerobinen Glykolyysivaiheen)
toisin toiminnon WT p53, mutantti p53-kasvaimen solujen suosii aerobinen glykolyysivaiheen, osittain parantamalla kaupan glukoosi transporter GLUT1 plasman kalvo, joten lisäämällä glukoosia tuonti (14, 15)., Seuraavat mutaatio p53, alennettua tasot SCO2 ja GLS2 ja kohonneeseen GLUT1 ja GLUT4 hyväksi aerobinen glykolyysivaiheen yli oksidatiivisen fosforylaation. Tällä tavalla, mutantti p53 uskotaan edistää taipumus kasvain solut käyttävät aerobinen glykolyysivaiheen hyväksi oksidatiivisen fosforylaation, tai ns Warburg aineenvaihduntaa (15). Yksi tunnusmerkkejä syöpä on vapautettu aineenvaihduntaa, yleensä osoituksena tämä vaihtaa aerobinen glykolyysivaiheen oksidatiivisen fosforylaation., Vaikka tämä johtaa vähemmän ja vähemmän tehokkaampi ATP: n tuotto, uskotaan, että syöpä solut hyötyä ohjaamalla glykolyyttisiä välituotteita, jotta biosynteettisiä reittejä välttämätöntä nopean solunjakautumisen (16). Tämä aineenvaihdunnan vaihtaa myös johtaa vähentynyt mitokondrioita-välitteisen apoptoosin ja tehokkaampaa signalointi kautta saatavilla aineenvaihduntatuotteiden syöpäsoluja (17).
Yhteinen Geneettinen Variantti, TP53 Vaikuttaa sen toimintaan Aineenvaihduntaa
on yleinen koodaus alueen polymorfismi p53 kodonissa 72, koodaus joko proline (P72) tai arginiini (R72)., Tämä aminohappojen vaihtelu voi vaikuttaa p53: n toimintaan solun kohtalon suhteen stressin jälkeen. Vastauksena DNA-vaurioita, P72 variantti p53 pääasiassa laukaisee solusyklin pidätys, kun R72 variantti pääasiassa aiheuttaa solun kuoleman, tai apoptoosin (18, 19). Huolimatta näistä eroista toiminto, kodonissa 72 vaihtelu ei ole ollut johdonmukaisesti liittyy syövän herkkyys (20). Sen sijaan ihmisillä tehdyissä tutkimuksissa tämä polymorfismi liittyy merkittävästi kohonneeseen painoindeksiin ja diabetesriskiin (21, 22)., Tämä lähtökohta tukee tutkimukset hiirillä, jossa hiiri malli näitä kodonissa 72 variantteja osoittaa lisääntynyt high-fat diet-induced diabetes hiirillä kanssa R72 variantti, verrattuna P72. Näissä tutkimuksissa p53-kohde-geenien TNFa: n ja NPC1L1 tunnistettiin kriittinen sääntelyviranomaisten lisätä ruokavalioon aiheuttama lihavuus R72 hiiret (23). Mielenkiintoista, R72 variantti on myös osoitettu antavan lisääntynyt selviytyminen solujen vastauksena ravinteiden puutteen (24)., Nämä havainnot ovat johtaneet hypoteesiin, että R72 variantti p53 nousi ja oli valittuna, kun väestö siirtyi pohjoiseen, jossa kylmä sää edellyttäisi, lisääntynyt rasvan kertymistä, mutta jossa selviytyminen vastauksena ravinteiden puutetta olisi myös alle valinta (24).
p53 Säätelee Rasva-Aineenvaihduntaa
Vaikka p53 on tunnettu säännellään glykolyysin ja sitruunahappokierron, p53 on myös osoitettu olevan rooli säännellä rasva-aineenvaihduntaa (25)., Uskotaan, että WT p53 parantaa rasvahappojen hapettumista estäen samalla rasvahappojen synteesiä ja toimii siten lipidisynteesin negatiivisena säätelijänä (25). On olemassa useita p53-kohdegeenejä, joilla on rooleja lipidimetaboliassa. Sanchez-Macedo ja työtovereiden osoittaneet, että karnitiini palmitoyltransferase 1C (CPT1C) on transcriptionally säännelty p53; tämä entsyymi auttaa liikenteen käytössä rasvahappojen mitokondriot., Tämän p53-säännellyn geenin roolin tukena syövässä tämä ryhmä osoitti, että Cpt1c-puutteellisilla hiirillä on viivästynyt kasvaimen kehitys ja korkeampi eloonjäämisaste (26). Lipin 1 (LPIN1) on toinen p53 kohde geeni; LPIN1 on tarpeen asianmukaisen adiposyyttien kehitystä ja on aiheuttama alhainen ravinteiden ehtoja (27). Finck ja kollegat osoittivat, että LPIN1 vuorovaikutuksessa PGC-1α, toisen tunnetun p53-kohde-geenin rooli aineenvaihduntaa, ja että tämä vuorovaikutus aktivoi geenien mukana on edistää rasvahappojen hapettumista (28).,
lisäksi suoraan säännellä transkriptio geenien mukana rasva-aineenvaihduntaa, p53 voi myös säädellä rasva-aineenvaihduntaa tavalla, johon suoraa proteiini–proteiini vuorovaikutus. Esimerkiksi, glukoosi-6-fosfaatti-dehydrogenaasi, joka on nopeutta rajoittava entsyymi pentoosifosfaattireitin, sitoo ja on suoraan estää p53, mikä laski NADPH tuotantoa ja näin ollen laski rasvahappojen synteesi (12)., Sterolien sääntelyn osa-sitovia proteiineja (SREBP) perheen transkriptiotekijät ohjaavat geenien mukana kolesterolia, rasvahappoja, asemassa, ja fosfolipidien synteesi (29-31). WT p53 sortaa SREBP-toiminto (32), kun taas mutantti muotoja p53 sitoa suoraan SREBP ja parantaa niiden transkription toimintaa, mikä lisää SREBP toimintaa ihmisen kasvaimia (33, 34). Näin ollen mutantti p53 korreloi sterolin biosynteesigeenien korkeamman ilmentymisen kanssa ihmisen rintojen kasvaimissa (34, 35)., Lopuksi, AMP-aktivoitu proteiinikinaasin (AMPK) on entsyymi, joka aktivoituu, alle alhainen ravinne-tai energia-stressiä, ja on tunnettua estää rasvahappojen synteesiä vuorovaikutuksessa asetyyli-CoA-carboxylase ja SREBP-1 (36, 37). Zhou ja kollegat osoittivat, että mutantti p53 ensisijaisesti sitoutuu ja estää AMPK, mikä lisää rasvahappojen synteesiä. Tämän seurauksena mutantti p53-proteiinit johtavat AMPK-signaloinnin lisääntymiseen, mikä edistää kasvainsolujen invasiivista solujen kasvua (33). Vähemmän tutkittu alue on p53: n rooli lipidikuljetuksissa., Se on ollut osoittaneet, että p53-transcriptionally säätelee apolipoproteiini B (apoB) ja apoB muokkaaminen entsyymi monimutkainen 1, mikä osoittaa, rooli p53 säätelyssä aterogeeniseen lipoproteiinien (38). Microarray-analyysi ihmisen maksan-johdettu solut tunnistetaan fosfolipidin siirtäjäproteiinin, ATP-sitova kasetti A12, ja karboksyyli esteri lipaasi kuin kolme p53 kohde-geenien, jotka kaikki pelata rooli rasva-liikenne (39, 40)., Kaiken kaikkiaan, vaikka se on selvää, että p53 on keskeinen rooli välittäjänä rasva synteesi ja aineenvaihdunta, vaikutus tämän reitin, ja nämä p53 kohde-geenien, kasvaimen tukahduttaminen p53 on vielä määriteltävä (Kuva 1).
Ferroptosis on Romaani Solun Kuoleman Polku ohjaa lipidiperoksidaation
Vuonna 2012, Dixon ja kollegat havaitsivat, romaanin muoto säädelty solukuolema kutsutaan ferroptosis. Ferroptoosi on raudasta riippuvainen, kaspaasista riippumaton solukuoleman muoto, joka johtuu hapettuneiden lipidien kertymisestä (4, 41)., Tämä prosessi ohjaa inaktivointi glutationi peroksidaasi 4 (GPX4), entsyymiä, joka on vastuussa muuntaa tappava lipid hydroperoxides myrkytön rasva-alkoholit, joka vaatii glutationi toimiakseen (41). Monityydyttymättömien rasvahappojen peroksidaation (PUFAs) uskotaan vauhdittavan ferroptoosin aiheuttamaa solukuolemaa. Pufa: sisältää bis-allylic protonit, jotka voidaan helposti hajamielinen ja tuottaa radikaaleja, jotka reagoivat hapen kanssa, luoda enemmän radikaaleja ja aiheuttaa ketjureaktion, rasva-reaktiivinen hapen lajien (42)., Tarkkaa mekanismia solun kuoleman ferroptosis on edelleen tuntematon, mutta yksi hypoteesi on, että rasva-vahingot johtaa tuhoaminen solukalvon (43). On arveltu, että ferroptosis voisi olla mekanismi kasvain tukahduttaminen, joka toimii poistamalla solut, jotka ovat ravinteiden puutteessa tai ovat altistuneet ympäristön stressiä tai infektio.,
Farmakologiset Asetus Ferroptosis
Ferroptosis voi olla aiheuttamaa käyttämällä estäjät järjestelmän xc− kuten erastin, tai analogit, kuten glutamaatin ja sorafenibi, jotka estävät tuonti kystiini, jolloin köyhdytettyä glutationi ja myöhemmin inaktivointi GPX4. Vaihtoehtoisesti, ferroptosis voi indusoida (1S,3R)-RSL3 (jäljempänä RSL3), joka suoraan sitoo ja estää GPX4 (4, 5, 42). Butionisulfoksimiini, FIN56, FINO2, CCl4 ja sisplatiini ovat muita aineita, joiden on osoitettu indusoivan ferroptoosia soluissa., Kuolema ferroptosis voidaan ehkäistä estämällä rasva-peroxidation, joka voidaan toteuttaa käyttämällä lipofiiliset antioksidantit, kuten ferrostatin-1, liproxstatin-1 tai E-vitamiinia, Rautaa kelaattorit, kuten deferoksamiini tai cicloprox ovat toinen väline, jota käytetään tukahduttaa ferroptosis vähentämällä rauta-arvot. Kufojen tyhjentäminen tai kertatyydyttymättömien rasvahappojen lisääminen soluviljelmiin voi myös pelastaa soluja ferroptoosilta (42, 44).,
Ferroptosis on Osallisena p53-Välitteisen Kasvain Tukahduttaminen
Vuonna 2012, Gu kollegoineen on kehittänyt hiiri malli, jossa kolme yleensä asetyloitu lysiini jäämiä DNA-binding domain p53 oli mutatoitunut arginiini, ja siksi voi olla asetyloitu; tämä hiiri on tarkoitettu, kuten 3KR hiiri. Erityisesti soluja 3KR hiiri eivät kykene läpikäymään p53-riippuvainen apoptoosin, solusyklin pidätys, tai vanhenemisen merkkejä, ja itse asiassa 3KR mutantti p53 ei transactivate suurin osa p53 kohde geenit., Kiinnostavaa kyllä, tämä hiiri malli ei spontaanisti kehittyä syöpä, mikä tarkoittaa, että p53 voi tukahduttaa kasvaimen kehitystä riippumaton vanheneminen tai apoptoosin (45). Tämä ryhmä totesi, että mutantti 3KR proteiini säilyy kyky suorittaa ferroptosis ja säädellä kystiini aineenvaihduntaa säätelemällä ilmaus kystiini maahantuoja SLC7A11; tämä ehdotti, että ferroptosis voisi olla yksi polku, joka taustalla p53-välitteisen kasvain tukahduttaminen., Kun villi tyyppi ja 3KR MEFs hoidettiin ferroptosis induktori Erastin, lähes 50% solun kuolema oli havaittu, että p53-null MEFs esillä 20% solun kuolema; tämä osoittaa, että p53 herkistää ihmisen solujen ferroptosis, ja myös, että muita keskeisiä sääntelyviranomaisten myös rooli ferroptosis (5). Myöhemmin, Gu ja työtovereiden tunnistaa ylimääräinen asetylointi-sivuilla osoitteessa lysiini 98, p53, ja ne syntyy hiiri malli, jossa kaikki neljä asetylointi sivustot olivat mutatoitunut arginiini (4KR)., Mielenkiintoista, 4KR mutantti oli mahdotonta säännellä geenien ferroptosis kuten SLC7A11, ja toisin kuin 3KR mutantti ei voinut tukahduttaa kasvaimen kehitystä (46). Vaikka tällä hetkellä kiinnekohta, nämä tiedot sekaantumaan rooli p53 vuonna ferroptosis sen kyky tukahduttaa kasvaimen kehitystä.
Ei-Transformoituja Soluja, p53 Säätelee Positiivisesti Ferroptosis
lisäksi SLC7A11, useita muita suoria p53 kohde geenejä on löydetty rooli ferroptosis. Näitä ovat esimerkiksi GLS2, PTGS2 ja SAT1., Kahdesta erillisestä ryhmästä tehdyt tutkimukset tukevat GLS2: n roolia ferroptoosissa, jonka tiedetään vähentävän glutationia ja lisäävän solujen ROS-tasoja. Jiang ja työtovereiden käyttää ferroptosis estäjät yhdistettynä glutaminolysis estäjät estää Erastin aiheuttama ferroptosis, mikä osoittaa, että ferroptosis vaatii glutaminolysis ja GLS2 (47). Murphy ja kollegat osoittivat, että polymorfinen variantti p53 pystyi aiheuttaa kasvun pysähtymisen ja vanheneminen sekä ihmisen ja hiiren soluissa, mutta ei tukahduttaa SLC7A11 tai transactivate GLS2., Tämä vaihtoehto oli selvästi alentunut asiakkuutta ferroptosis ja tukahduttaa kasvaimen kehitystä, mikä taas sotki rooli p53 vuonna ferroptosis-välitteisen kasvain tukahduttaminen (48). Toinen p53-kohdegeeni, jolla on rooli ferroptoosissa, on ptgs2, syklo-oksigenaasi-2-entsyymiä koodaava geeni. Stockwell ja kollegat osoittivat ensimmäistä kertaa, että induktio ferroptosis käyttäen Erastin ja RSL3 johti säätelyä PTGS2 (41). Erityisesti, PTGS2 ei upregulated, jonka ferroptosis induktorit in p53-null solut, mikä viittaa siihen, että tämä asetus on riippuvainen p53 (5)., Nykyisin PTGS2: n säätelyä käytetään laajalti ferroptoosimerkkinä (5, 41).
Gu-ryhmän tuore tutkimus osoitti, että p53-kohdegeeni SAT1 säätelee ferroptoosia (49). Kirjoittajat tunnistaa SAT1 kuin suora tavoite p53 ja osoitti, että vaientaminen SAT1 vähentää solun kuoleman aiheuttama reaktiivinen hapen lajien solujen WT p53, mutta ei ollut vaikutusta p53-null solut. Mekaanisesti tämä ryhmä osoitti, että SAT1 lisää taso ja aktiivisuus arachidonate 15-lipoxygenase, rauta-sitovia entsyymi, joka hapettaa Pufa ja lisää rasva-peroxidation., Tämä tutkimus osoitti erityisesti, että pelkkä p53 tai SAT1 ei näytä olevan riittävä aiheuttamaan ferroptoosia. Sen sijaan, yhdistetty tiedot ovat sopusoinnussa oletuksesta, että p53, nojalla sääteleviä geenejä, jotka edistävät ferroptosis, säätelee solujen herkkyys tämän reitin, eikä suoraan indusoi ferroptosis. Se, sääteleekö p53 muita ferroptoosiin osallistuvia geenejä, jää selvittämättä (kuva 2).
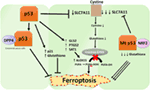
Kuva 2. P53: n eri roolit ferroptoosissa., Esto glutationi peroksidaasi 4 (GPX4), keskeinen entsyymi, joka katalysoi monityydyttymättömiä rasvahappoja (Pufa), jotka sisältävät peroksideja, jotta alkoholit, on avain kuljettaja ferroptosis. Kontekstista riippuen p53 voi tukahduttaa ferroptoosia (kuten kolorektaalisyöpäsoluissa) tai edistää ferroptoosia. Mutantti p53 herkistää soluja ferroptoosille jopa enemmän kuin villityypin p53.,
Joissakin Soluissa, p53 Säätelee Negatiivisesti Ferroptosis
äskettäin julkaiseman tutkimuksen Tarangelo ja työtovereiden osoittavat, että p53 säätelee negatiivisesti ferroptosis syövän solut (50). Tämä ryhmä havaitsi, että p53: a stabiloivan Nutlin-3: n esikäsittelevät solut viivästyttävät ferroptoosin puhkeamista useissa solutyypeissä. Viivästynyt puhkeamista ferroptosis löytyi riippuu CDKN1A (koodaus p21), kriittinen p53 transkription kohde., Mekanismi, jonka kautta p21 viivästyksiä ferroptosis ei ole vielä selvitetty, mutta uskotaan, että säilyttämistä solunsisäisen glutationin saattaa olla myötävaikuttava tekijä vähentää ferroptosis herkkyys. Kirjoittajat toteavat, että p53–p21-akseli mahdollistaa syöpäsolujen hengissä olosuhteissa metabolinen stressi, kuten kystiini puutetta, tukahduttamalla puhkeamista ferroptosis (50). Tuore tutkimus osoitti, että p53 estää ferroptosis paksu-ja peräsuolen syövän soluja sitoutumalla entsyymin alentavat-peptidaasi-4: n (DPP4), joka on modulaattori ferroptosis-ja rasva-aineenvaihduntaa., Mekaanisesti tämä tutkimus osoitti, että p53 estää ferroptosis, jonka eristävät DPP4 vuonna ydinvoima entsymaattisesti aktiivinen-allas. P53: n puuttuessa DPP4 on vapaa vuorovaikutuksessa ja muodostamaan kompleksin NOX1: n kanssa; tämä johtaa lisääntyneeseen lipidiperoksidaatioon ja ferroptoosiin. Esto DPP4 estää ferroptosis merkittävästi, kun taas yli-ilmentymä DPP4 laukaisee Erastin herkkyys, erityisesti p53-köyhdytettyä solut (51). Kaksisuuntainen ohjaus ferroptosis, jonka kautta p53 transkription-riippuvainen ja transkriptio-riippumattomat mekanismit voivat olla yhteydessä tai solu-tyyppi riippuvainen (Kuva 2).,
The P47S Polymorfismi TP53 Vaikuttaa Ferroptosis ja Kasvain Tukahduttaminen
lisäksi näin mutaatioita, on olemassa useita toiminnallisesti merkittäviä yhden nukleotidin polymorfismien (snp) vuonna TP53-geenin ja muiden proteiinien tiedetään säännellä tämän reitin (kuten MDM2 ja MDM4). Se Pro47Ser variant (jäljempänä S47) on toiseksi yleisin SNP löytyy p53-koodaava alue (sen jälkeen, kun Pro72Arg), joka muuttaa aminohapon sekvenssi proteiinia., Paremmin valaista vaikutus tämän muunnelma p53 toiminta ja syövän riski, Murphy ryhmä syntyy humanisoitu p53 knock-hiiren malli, jossa eksonien 4-9 hiiren p53 korvattiin ihmisen p53-eksonien, jossa on joko villi tyyppi tai S47 variant (52-55). Suurin osa S47-hiiristä kehitti spontaanisti erilaisia histologisia tyyppejä, erityisesti maksasyöpää, sisältäviä kasvaimia 12-18 kuukauden ikäisinä, toisin kuin WT p53-hiirillä (48)., Hiiren alkion fibroblastit ja ihmisen lymphoblastoid solu linjat, S47 variantti osoitti heikentynyt ohjelmoidun solukuoleman vastauksena sisplatiinin ja muita genotoksisia korostaa. Mekanistisesti S47-muunnos on puutteellinen aineenvaihduntaan osallistuvien geenien, kuten Gls2: n (glutaminaasi 2) ja Sco2: n (48) transaktivoinnissa. Yhdenmukainen rooli Gls2 vuonna ferroptosis, tämä ryhmä totesi, että S47 solut olivat selvästi kestävät ferroptosis aiheuttavia aineita Erastin ja RSL3 (47, 48). Tämä vika voi edistää S47-hiirillä havaittua kasvainherkkää fenotyyppiä.,
Mutant p53 herkistää ihmisen Kasvain Solujen Ferroptosis
Villi-tyyppi p53 säätelee negatiivisesti ilmaus kystiini maahantuoja SLC7A11, joka estää herkkyys ferroptosis (5). Vaikka tämä asetus tapahtuu normaalien solujen, kasvain soluja, muita välittäjiä SLC7A11 näyttävät enemmistönä asetuksen tämän geenin. Esimerkiksi master antioksidantti transkriptio tekijä NRF2 voi myös säädellä ilmaus SLC7A11 klo transkription tasolla, ja NRF2 on ollut mukana keskeisenä toimijana suojella syöpäsoluja vastaan ferroptosis., Esimerkiksi esto NRF2 vuonna hepatosellulaarinen syöpä soluja lisää anti-syöpä toimintaa Erastin ja Sorafenibi in vivo (56). Mutant muotoja p53 voi estää NRF2-toiminto suoraa vuorovaikutusta, ja yksi ryhmä totesi, että kasvaimia mutant p53 sisältävät hyvin alhainen SLC7A11, ja siten osoittavat lisääntynyt herkkyys ferroptosis. Erityisesti, yli-ilmentymä SLC7A11 mutant p53-mallit led huumeiden vastarintaa, mikä viittaa siihen, että tasot SLC7A11 ilme on otettava huomioon, kun kohdistaminen mutantti p53 ajaa syövät ferroptosis aiheuttavia yhdisteitä (57)., Tämän tueksi lähtökohta, viime työtä kolorektaalisyövän (CRC) syöpä, jossa mutaatio tai deleetio p53 on usein tapahtuma, osoitti, että ihmisen CRC solulinjoja kätkeminen mutantti p53 olivat paljon herkempiä Erastin-välitteisen solukuoleman, kun sitä verrataan CRC-solujen WT p53. Näiden havaintojen validoimiseksi ne osoittivat, että sekä HCT116-että SW48-soluissa tapahtunut p53-hotspot-mutaatio palautti herkkyyden Erastiinille (51). Nämä tiedot korostavat uudenlaista mekanismia, jonka avulla mutantti p53: n ajamia syöpiä voidaan hyödyntää kohdennetulla hoidolla.,
Johtopäätös
rooli p53-aineenvaihdunnassa on aivan selvää, ja mahdollisesti jopa intuitiivisesti ilmeinen: WT p53 rajoittaa glukoosin aineenvaihduntaa ja rasva synteesi, kun mutantti p53 tulee tehdä päinvastoin. Panos sen aineenvaihdunnan rooli syövän tukahduttaminen p53, ja kyky mutantti p53 ajaa kasvaimen etenemiseen, jää yksiselitteisesti todistettu. Rooli p53-asetuksessa ferroptosis, ja osuus tämä toiminto, kasvain tukahduttaminen on vielä vähemmän selvää., Kun taas pakottavia tietoja hiiren malleja tukee oletuksesta, että p53 säätelee herkkyyttä solujen ferroptosis, tämä voi olla rajoitettu kyky pohjapinta p53 tukahduttaa spontaani kasvaimen kehitystä, ja onkogeeni-korosti hiiri malleja, on selvää, että vanheneminen ja apoptoosin pelata hallitseva asema. Vastaavasti p53 voi säädellä ferroptoosin herkkyyttä solutyyppikohtaisesti. Lisää tutkimuksia eläinmalleissa, kiinnittäen huomiota ferroptoosiin eri kudoksissa, on tehtävä, jotta voidaan paremmin ymmärtää p53: n rooli ferroptoosissa ja ferroptoosissa kasvaimen tukahduttamisessa., Lisäksi selkeämpi käsitys siitä, mitä p53-kohde-geenien rooli herkkyys ferroptosis on tarkoitus saavuttaa. Näiden kysymysten ratkaisemisen pitäisi tarjota kaivattuja uusia keinoja torjua kasvaimia mutantti p53: lla.
Kirjoittaja Maksut
KG, SB -, TB -, AB -, K -, C-PK, ja MM jokainen kirjoitti kaksi kohdissa tämän artikkelin. KG ja SB tekivät luvun. KG ja MM hahmottelivat luvun.,
eturistiriita Lausunto
kirjoittajat ilmoittavat, että tutkimus on tehty ilman mitään kaupallisia tai taloudellisia suhteita, jotka voitaisiin tulkita mahdollisia eturistiriitoja.
arvostelija OAF ja käsittelytoimittaja ilmoittivat yhteenliittymisestään.
Kiitokset
Tutkimus raportoitu tässä julkaisussa tukivat National Institutes of Health nojalla Palkinto Numerot CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK), ja T32 CA009171 (TB)., Sisältö on yksinomaan tekijöiden vastuulla, eikä se välttämättä edusta kansallisten Terveysinstituuttien virallisia näkemyksiä.
3. Humpton TJ, Vousden KH. Soluaineenvaihdunnan ja hypoksian säätely p53: lla. Cold Spring Harb Perspect Med (2016) 6(7):211-30. doi:10.1101 / cshperspect.a026146
CrossRef Koko Teksti | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., Tuumorisuppressori p53 säätelee negatiivisesti hypoksian stimuloimaa glykolyysiä kohteensa RRAD kautta. Oncotarget (2014) 5(14):5535-46. doi:10.18632 / oncotarget.2137
PubMed Abstrakti | CrossRef Koko Teksti | Google Scholar
17. Lee M, Yoon JH. Glykolyysin ja mitokondrioiden hapettumisen välinen metabolinen vuorovaikutus: Käänteinen Warburgin vaikutus ja sen terapeuttinen vaikutus. World J Biol Chem (2015) 6(3):148-61. doi:10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., Uuden p53-target-geenin karnitiinipalmitoyylitransferaasi 1C: n ehtyminen viivästyttää kasvaimen kasvua neurofibromatoosin tyypin I kasvainmallissa. Solukuolemat Vaihtelevat (2013) 20(4):659-68. doi:10.1038 / cdd.2012.168
PubMed Abstrakti | CrossRef Koko Teksti | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave MINUA, et al. Dysregulaatio steroli-vaste-elementti-sitovia proteiineja ja loppupään mikseri eturauhassyövän etenemisen aikana androgeenireseptorin itsenäisyyttä., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong VM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-hiirillä on kimeerinen ihmisen ja hiiren p53-geenin kehittyä normaalisti ja näyttää villi-tyyppi p53 vastauksia DNA haitallisia aineita: uusi biolääketieteen tutkimuksen työkalu. Onkogeeni (2001) 20(3):320-8. doi:10.1038 / sj.onc.1204080
PubMed Abstrakti | CrossRef Koko Teksti | Google Scholar
57. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Järjestelmän XC-/glutationiakselin esto kohdistuu selektiivisesti syöpiin, joissa mutantti-p53 kumuloituu., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















