Frontiers in Endocrinology (Magyar)
Introduction
a tumorszuppresszor gén TP53 már a legtöbb erősen tanulmányozott emberi gén felfedezése óta közel 40 évvel ezelőtt (1). Ennek az állapotnak a fő oka a p53 kritikus szerepe a rák kialakulásának megelőzésében, amelyet széles körben “a genom őrzőjének” tekintnek.,”Egy ideje azt hitték, hogy a p53 szerepe a tumorszuppresszióban az apoptózis, a sejtciklus letartóztatásának és a rák előtti sejtek szeneszcenciájának indukálására képes (2). Most azonban egyre világosabbá válik, hogy a p53 a sejt számos más útvonalát szabályozza, és hogy ezek a más utak szerepet játszanak a p53 tumorszuppresszorként való működésében is (3). Különösen a p53 szerepe az anyagcserében és a ferroptózisban részt vevő gének szabályozásában szerepet játszott a tumor fejlődésének elnyomásában., A ferroptózis egy új sejthalál-útvonal, amelyet először 2012-ben jellemeztek, és legjobban a sejthalál vasfüggetlen, kaszpázfüggetlen formájaként írható le, amelyet a lipidperoxidáció kialakulása vezérel (4). Pontosabban, két egér modell tartalmaz mesterséges mutációk p53, hogy megszüntesse a képessége, p53, hogy rábírja a apoptózis, valamint senescence mindketten megtartják a képességgel, hogy elnyomja a spontán tumor fejlődését; mindkét mutánsok megtartja a képesség, hogy transactivate gének anyagcserét, valamint ferroptosis (5, 6)., A metabolizmus és a ferroptózis szabályozásában a p53-at befolyásoló adatok összefoglalása az alábbiakban található.
Vad Típusú (WT) p53 Pozitívan Szabályozza Oxidatív Foszforiláció, illetve Elnyomja a Glükóz Anyagcsere
Vad típusú p53 szabályozza az anyagcsere-sokoldalúság, a sejtek által előnyben mitokondriális légzés át glikolízis, részben keresztül a transactivation a SCO2 (citokróm c oxidáz közgyűlés), amelyet játszik közvetlen szerepet játszik az oxidatív foszforiláció (7)., a p53 közvetlenül szabályozza a GLS2 (Glutamináz 2) transzaktiválását is; ez az enzim lehetővé teszi a glutamin felhasználását a mitokondriumok energiaforrásaként (8). Ezenkívül a WT p53 negatívan szabályozza a glikolízist a glut1 és GLUT4 glükózszállítók transzkripciós elnyomásával, valamint a RRAD és a TIGAR transzaktiválásával; mindkettő glikolízis inhibitora (9-11). Végül a p53 közvetlenül kötődik és gátolja a glükóz-6-foszfát-dehidrogenáz enzimet, ezáltal gátolja a glükóz metabolizmusát (12)., Ezekből és más vizsgálatokból kitűnik, hogy normál, feszítetlen organizmusokban a p53 közvetlenül szabályozza a sejt metabolikus állapotát (1.ábra). Nem meglepő, hogy ez a gén és számos szabályozója szerepet játszik az anyagcsere-betegségekben, köztük az elhízásban és a cukorbetegségben (13).
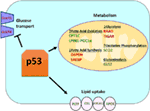
1.ábra. A vad típusú (WT) p53 szerepe az anyagcserében. A p53 által pozitívan szabályozott géneket zölden mutatják, a p53 által negatívan szabályozott géneket pedig pirosan mutatják., a p53 gátolja a glükóz transzportot, a glikolízist és a zsírsavszintézist, miközben elősegíti a lipid felvételét, a zsírsav oxidációját, az oxidatív foszforilációt és a glutaminolízist.
Mutáns p53 Pozitívan Szabályozza Warburg Anyagcsere (Aerob Glikolízis)
ezzel szemben a funkció a WT p53 mutáns p53 tumor sejtek szívességet aerob glikolízis, részben azáltal, hogy növeli a kereskedelem, a glükóz transzporter GLUT1, hogy a plazma membrán, ezért nő a glükóz import (14, 15)., A p53 mutációját követően az SCO2 és GLS2 csökkent szintje, valamint a GLUT1 és GLUT4 megnövekedett szintje elősegíti az aerob glikolízist az oxidatív foszforiláció felett. Ily módon úgy gondolják, hogy a mutáns p53 hozzájárul a tumorsejtek hajlandóságához, hogy aerob glikolízist használjanak az oxidatív foszforiláció vagy az úgynevezett Warburg metabolizmus (15) javára. A rák egyik jellemzője a deregulált anyagcsere, amelyet általában az aerob glikolízisről az oxidatív foszforilációra való átállás igazol., Bár ez alacsonyabb és kevésbé hatékony ATP-hozamot eredményez, úgy gondolják, hogy a rákos sejtek előnyösek azáltal, hogy a glikolitikus intermediereket a gyors sejtosztódáshoz szükséges bioszintetikus útvonalakra terelik (16). Ez a metabolikus kapcsoló a mitokondriumok által közvetített apoptózis csökkenéséhez és a rákos sejtekben rendelkezésre álló metabolitok révén történő hatékonyabb jelátvitelhez is vezet (17).
A TP53 közös genetikai változata befolyásolja a metabolizmus funkcióját
a p53 közös kódolási régió polimorfizmusa a 72 kodonban, kódolva a prolin (P72) vagy az arginin (R72) számára., Ez az aminosav-variáció hatással lehet a p53 funkcióra a sejtek sorsára a stressz után. A DNS-károsodásra válaszul a p53 P72 változata túlnyomórészt sejtciklus-letartóztatást vált ki, míg az R72 változat túlnyomórészt sejthalálot vagy apoptózist (18, 19) okoz. A funkcióbeli különbségek ellenére a codon 72 variációja nem volt következetesen összefüggésben a rák érzékenységével (20). Ezzel szemben az emberi vizsgálatokban ez a polimorfizmus jelentősen összefügg a testtömeg-index növekedésével és a cukorbetegség kockázatával (21, 22)., Ezt a feltevést egereken végzett vizsgálatok támasztják alá, ahol ezeknek a kodon 72 változatoknak az egérmodellje az R72 változattal rendelkező egerekben a magas zsírtartalmú étrend által kiváltott cukorbetegséget mutatja, a P72-hez képest. Ezekben a vizsgálatokban a TNFa és az NPC1L1 p53 célgéneket kritikus szabályozóként azonosították az étrend által kiváltott elhízás növekedésében R72 egerekben (23). Érdekes módon az R72 változatról azt is kimutatták, hogy növeli a sejtek túlélését a tápanyaghiányra adott válaszként (24)., Ezek az eredmények azt a hipotézist eredményezték, hogy a p53 R72 változata felmerült, és észak felé vándoroltak, ahol a hideg időjárás fokozott zsírfelhalmozódást igényelne, de ahol a tápanyaghiányra adott válaszként a túlélés is kiválasztás alatt állna (24).
p53 szabályozza a Lipid anyagcserét
bár a p53 jól ismert a glikolízis és a citromsav ciklus szabályozásában, a p53 szintén szerepet játszott a lipid metabolizmus szabályozásában (25)., Úgy gondolják, hogy a WT p53 fokozza a zsírsav oxidációját, miközben gátolja a zsírsav szintézisét, így a lipidszintézis negatív szabályozójaként működik (25). Számos p53 célgén van, amelyek szerepet játszanak a lipid anyagcserében. Sanchez-Macedo és munkatársai bebizonyították, hogy a karnitin-palmitoil-transzferáz 1C (CPT1C) transzkripcionálisan szabályozza a p53; ez az enzim segíti az aktivált zsírsavak szállítását a mitokondriumokba., Ennek a p53-szabályozott génnek a rákban betöltött szerepének alátámasztására ez a csoport kimutatta, hogy a Cpt1c-hiányos egerek késleltetett tumorfejlődést és magasabb túlélési arányt mutatnak (26). A Lipin 1 (LPIN1) egy másik p53 célgén; az lpin1 szükséges a megfelelő adipocita fejlődéshez, és alacsony tápanyag-körülmények között indukálódik (27). Finck és kollégái kimutatták, hogy az lpin1 kölcsönhatásba lép a PGC-1α-val, egy másik ismert p53 célgénnel, amely szerepet játszik az anyagcserében, és hogy ez a kölcsönhatás aktiválja a zsírsav-oxidáció elősegítésében részt vevő gének expresszióját (28).,
amellett, hogy közvetlenül szabályozza a lipid anyagcserében részt vevő gének transzkripcióját, a p53 a lipid anyagcserét közvetlen fehérje–fehérje kölcsönhatással járó módon is szabályozhatja. Például a glükóz-6-foszfát-dehidrogenáz, amely a pentóz-foszfát út sebességkorlátozó enzimje, kötődik a p53-hoz, és közvetlenül gátolja, ami csökkent NADPH termelést, következésképpen csökkent zsírsavszintézist eredményez (12)., A transzkripciós faktorok szterol-szabályozó elem-kötő fehérjék (SREBP) családja modulálja a koleszterin, a zsírsav, a triacil-glicerin és a foszfolipid szintézisben részt vevő gének expresszióját (29-31). A WT p53 elnyomja a SREBP funkciót (32), míg a p53 mutáns formái közvetlenül a SREBP-hez kötődnek, és fokozzák transzkripciós funkciójukat, ami fokozott SREBP aktivitást eredményez az emberi daganatokban (33, 34). Következésképpen a mutáns p53 korrelál a szterin bioszintézis gének magasabb expressziójával az emberi emlődaganatokban (34, 35)., Végül, az AMP-aktivált protein kináz (AMPK) egy olyan enzim, amely alacsony tápanyagszint vagy energia stressz hatására aktiválódik, és ismert, hogy gátolja a zsírsav szintézisét az acetil-CoA-karboxilázzal és a SREBP-1-vel (36, 37) való kölcsönhatás révén. Zhou és munkatársai kimutatták, hogy a mutáns p53 előnyösen kötődik és gátolja az AMPK-t, ami fokozott zsírsavszintézist eredményez. Ennek eredményeként a mutáns p53 fehérjék fokozott AMPK jelátvitelt eredményeznek, hozzájárulva a tumorsejtek invazív sejtnövekedéséhez (33). Kevésbé feltárt terület a p53 szerepe a lipidszállításban., Kimutatták, hogy a p53 transzkripcionálisan szabályozza az apolipoprotein B (apoB) és az apoB 1 enzimkomplexet, jelezve a p53 szerepét az aterogén lipoproteinek szabályozásában (38). Az emberi májból származó sejtek mikroarray elemzése a foszfolipid transzferfehérjét, az ATP A12 kötőkazettát és a karboxil-észter lipázt három p53 célgént azonosította, amelyek mind szerepet játszanak a lipidszállításban (39, 40)., Összességében, bár egyértelmű, hogy a p53 kulcsszerepet játszik a lipidszintézis és az anyagcsere közvetítésében, ennek az útvonalnak és ezeknek a p53 célgéneknek a hozzájárulása a p53 tumorszuppressziójához még meg kell határozni (1.ábra).
A Ferroptózis egy új sejthalál útvonal, amelyet Lipid peroxidáció vezet
2012 – ben Dixon és kollégái felfedezték a szabályozott sejthalál új formáját, a ferroptózist. A ferroptózis az oxidált lipidek (4, 41) felhalmozódásából eredő sejthalál vasfüggő, kaszpázfüggetlen formája., Ezt a folyamatot a glutation-peroxidáz 4 (GPX4) inaktiválása hajtja végre, amely enzim felelős a halálos lipid-hidroperoxidok nem toxikus lipid-alkoholokká történő átalakításáért, amely működéséhez glutationra van szükség (41). Úgy gondolják, hogy a többszörösen telítetlen zsírsavak (PUFA-k) peroxidációja a ferroptózis által okozott sejthalál hajtóereje. A PUFA-k olyan bisz-allil protonokat tartalmaznak, amelyek könnyen kivonhatók, és olyan gyököket termelnek, amelyek oxigénnel reagálnak, több gyököt hoznak létre és lipid reaktív oxigén fajok láncreakcióját eredményezik (42)., A sejthalál pontos mechanizmusa ferroptózissal továbbra sem ismert, de az egyik hipotézis az, hogy a lipidkárosodás a plazmamembrán megsemmisítéséhez vezet (43). Feltételezték, hogy a ferroptózis a tumorszuppresszió mechanizmusa lehet, amely úgy működik, hogy eltávolítja a tápanyagtól megfosztott vagy környezeti stressznek vagy fertőzésnek kitett sejteket.,
A Ferroptózis farmakológiai szabályozása
Ferroptózis indukálható XC rendszer− például erasztin-inhibitorokkal vagy analógokkal, például glutatinnal és szorafenibbel, amelyek gátolják a cisztin behozatalát, ami a glutation kimerülését és a gpx4 későbbi inaktiválódását eredményezi. Alternatív megoldásként a ferroptózist (1S,3R)-RSL3 (a továbbiakban: RSL3) indukálhatja, amely közvetlenül kötődik a GPX4-hez (4, 5, 42) és gátolja. A bution-szulfoximin, a FIN56, a FINO2, a CCl4 és a ciszplatin más olyan szerek, amelyekről kimutatták, hogy ferroptózist indukálnak a sejtekben., A ferroptózis okozta halál megelőzhető a lipidperoxidáció elnyomásával, amelyet lipofil antioxidánsok, például ferrosztatin-1, lipoxstatin-1 vagy E-vitamin alkalmazásával lehet elérni.Vaskelátorok, például deferoxamin vagy cicloprox egy másik eszköz a ferroptózis elnyomására a vas szintjének csökkentésével. A Pufák lebontása vagy az egyszeresen telítetlen zsírsavak hozzáadása a sejttenyészet közegéhez szintén megmentheti a sejteket a ferroptózistól (42, 44).,
Ferroptosis az ügyben, a p53 által Közvetített Daganat Elnyomás
2012-Ben, Gu, a kollégák pedig kifejlesztett egy egér modell, amelyben három rendszerint acetilezett lizin maradékok a DNS-kötő domén a p53 volt mutáns, hogy arginin, ezért nem lehet acetilezett; ez az egér a továbbiakban a 3KR egér. Nevezetesen, a 3kr egér sejtjei nem képesek p53-függő apoptózison, sejtciklus-letartóztatáson vagy szeneszcencián átesni, sőt a p53 3kr mutánsa nem képes transzaktiválni a p53 célgének többségét., Érdekes, hogy ez az egér a modell nem spontán alakul ki a rák, utalva arra, hogy a p53 lehet elnyomni a tumor fejlődését független senescence vagy apoptózis (45). Ez a csoport megállapította, hogy a mutáns 3kr fehérje megtartja a képességét, hogy részt ferroptosis és szabályozza cisztin anyagcsere szabályozásával kifejezés a cisztin importőr SLC7A11; ez arra utal, hogy a ferroptózis lehet az egyik út, amely alapja a p53-mediált tumor elnyomása., Amikor a vad típust és a 3kr MEFs-t ferroptosis induktor Erasztinnel kezelték, közel 50% sejthalált figyeltek meg, míg a p53 null MEFs 20% sejthalált mutatott; ez azt jelzi, hogy a p53 szenzibilizálja a sejteket ferroptózisra, valamint hogy más kulcsfontosságú szabályozók is szerepet játszanak a ferroptózisban (5). Ezt követően a Gu és munkatársai további acetilációs helyet azonosítottak a p53 lizin 98-ban, és létrehoztak egy egérmodellt, amelyben mind a négy acetilációs helyet argininre (4KR) mutálták., Érdekes módon a 4KR mutáns nem tudta szabályozni a ferroptózisban részt vevő géneket, mint például az SLC7A11, és a 3kr mutánssal ellentétben nem tudta elnyomni a tumor fejlődését (46). Bár jelenleg korrelatív, ezek az adatok magukban foglalják a p53 szerepét a ferroptózisban a tumor fejlődésének elnyomásában.
nem transzformált sejtekben a p53 pozitívan szabályozza a Ferroptózist
az SLC7A11 mellett számos más közvetlen p53 célgént fedeztek fel, amelyek szerepet játszanak a ferroptózisban. Ezek közé tartozik a GLS2, a PTGS2 és a SAT1., Két különböző csoportból származó vizsgálatok alátámasztják a GLS2 szerepét a ferroptózisban, amelyről ismert, hogy csökkenti a glutationszintet és növeli a sejtes ROS szintet. Jiang és munkatársai glutaminolízis inhibitorokkal kombinált ferroptózis inhibitorokat alkalmaztak az Erasztin által indukált ferroptózis gátlására, ezáltal bizonyítva, hogy a ferroptózis glutaminolízist és GLS2-t igényel (47). Murphy és kollégái azt mutatták, hogy a p53 polimorf változata képes volt a növekedés megállítására és a szeneszcencia kiváltására mind az emberi, mind a Murin sejtekben, de nem sikerült elnyomni az SLC7A11-et vagy a transzaktivált GLS2-t., Ez a változat jelentősen csökkent a ferroptózis indukálásakor és a tumor fejlődésének elnyomásakor, így ismét a p53 szerepét vonta maga után a ferroptózis által közvetített tumorszuppresszióban (48). Egy másik p53 célgén, amely szerepet játszik a ferroptózisban, a PTGS2, a ciklooxigenáz-2 enzimet kódoló gén. Stockwell és kollégái először azt mutatták, hogy az erastin és RSL3 alkalmazásával történő ferroptózis indukciója a PTGS2 (41) szabályozásához vezetett. Nevezetesen, a p53-null sejtekben a ferroptózis induktorok nem szabályozták a PTGS2-t, ami arra utal, hogy ez a rendelet p53-függő (5)., Jelenleg a PTGS2 upregulációját széles körben használják ferroptózis markerként (5, 41).
A Gu csoport egy nemrégiben végzett tanulmánya kimutatta, hogy a p53 CÉLGÉN, a SAT1 szabályozza a ferroptózist (49). A szerzők azonosított SAT1, mint a közvetlen cél a p53 pedig azt mutatta, hogy a hangtompító a SAT1 csökken a sejt halálát okozta reaktív oxigén fajok sejtek WT p53, de nem volt hatása a p53-null sejtek. Mechanikusan ez a csoport azt mutatta, hogy a SAT1 növeli az arachidonát 15-lipoxigenáz szintjét és aktivitását, egy vaskötő enzimet, amely oxidálja a Pufákat és növeli a lipidperoxidációt., Nevezetesen, ez a tanulmány kimutatta, hogy sem a p53, sem a SAT1 önmagában nem tűnik elegendőnek a ferroptózis kiváltásához. Ehelyett a kombinált adatok jobban összhangban vannak azzal a feltevéssel, hogy a p53 a ferroptózishoz hozzájáruló gének szabályozásával szabályozza a sejtek érzékenységét erre az útra, nem pedig közvetlenül indukálja a ferroptózist. Meg kell határozni, hogy a p53 szabályozza-e a ferroptózisban részt vevő egyéb géneket (2.ábra).
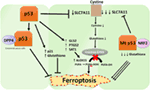
2.ábra. A p53 különböző szerepei a ferroptózisban., A glutation-peroxidáz 4 (GPX4) gátlása, a peroxidokat tartalmazó többszörösen telítetlen zsírsavak (Pufák) alkoholokká történő átalakulását katalizáló kulcsfontosságú enzim, a ferroptózis fő hajtóereje. A kontextustól függően a p53 elnyomhatja a ferroptózist (például a colorectalis rákos sejtekben), vagy elősegítheti a ferroptózist. A mutáns p53 még jobban érzékenyíti a sejteket a ferroptózisra, mint a vad típusú p53.,
egyes sejtekben a p53 negatívan szabályozza a Ferroptózist
a Tarangelo és kollégái által nemrégiben közzétett tanulmány azt mutatja, hogy a p53 negatívan szabályozza a ferroptózist a rákos sejtekben (50). Ez a csoport azt találta, hogy a Nutlin-3-mal, a p53-at stabilizáló vegyülettel történő előkezelés késlelteti a ferroptózis kialakulását több sejttípusban. A ferroptózis késleltetett kialakulása a cdkn1a-tól (P21 kódolás) függ, amely kritikus p53 transzkripciós cél., A mechanizmus, amelyen keresztül a p21 késlelteti a ferroptózist, még nem tisztázott, de úgy gondolják, hogy az intracelluláris glutation megőrzése hozzájárulhat a csökkent ferroptózis érzékenységhez. A szerzők arra a következtetésre jutnak, hogy a p53–p21 tengely lehetővé teszi a rákos sejtek túlélését metabolikus stressz körülmények között, például cisztin nélkülözés mellett, a ferroptózis kialakulásának elnyomásával (50). Egy nemrégiben készült tanulmány kimutatta, hogy a p53 gátolja a ferroptózist a colorectalis rákos sejtekben a dipeptidil-peptidáz-4 (DPP4) enzimhez való kötődéssel, amely a ferroptózis és a lipid metabolizmus modulátora., Mechanikusan ez a tanulmány kimutatta, hogy a p53 antagonizálja a ferroptózist a dpp4 szekvesztálásával egy nukleáris enzimatikus inaktív medencében. P53 hiányában a DPP4 szabadon kölcsönhatásba léphet és komplexet képezhet az NOX1-gyel; ez fokozott lipidperoxidációhoz és ferroptózishoz vezet. A dpp4 gátlása jelentősen elnyomja a ferroptózist, míg a DPP4 túlzott expressziója Erasztin érzékenységet vált ki, különösen a p53-kimerült sejtekben (51). A ferroptózis kétirányú szabályozása p53-mal transzkripciófüggő és transzkripciófüggetlen mechanizmusokon keresztül összefüggésben vagy sejttípusfüggő lehet (2.ábra).,
A TP53 P47s polimorfizmusa befolyásolja a Ferroptózist és a Tumorszuppressziót
a missense mutációk mellett számos funkcionálisan jelentős egy nukleotid polimorfizmus (SNPs) van a TP53 génben, és más fehérjék, amelyekről ismert, hogy szabályozzák ezt az utat (például MDM2 és MDM4). A Pro47Ser változat (a továbbiakban: S47) a második leggyakoribb SNP a p53 kódolási régióban (Pro72Arg után), amely megváltoztatja a fehérje aminosav-szekvenciáját., Jobb tisztázni a hatását ez a változat a p53 funkció, valamint a rák kockázatát, a Murphy csoport létrehozott egy humanizált p53 knock-egér modell, amelyben exons 4-9 az egér p53 helyébe emberi p53 exons tartalmazó, vagy a vad típusú, vagy a S47 változat (52-55). Az S47 egerek többsége spontán módon, a WT p53 egerekkel (48) ellentétben, különböző szövettani típusú, különösen májrákos daganatokat fejlesztett ki 12 és 18 hónapos kor között., Egér embrionális fibroblasztokban és humán lymphoblastoid sejtvonalakban az S47 variáns ciszplatin és más genotoxikus stressz hatására károsodott programozott sejthalált mutatott. Mechanikusan az S47 változat hibás az anyagcserében részt vevő gének, például a Gls2 (glutamináz 2) és a Sco2 (48) transzaktiválására. A Gls2 ferroptózisban betöltött szerepével összhangban ez a csoport megállapította, hogy az S47 sejtek jelentősen ellenálltak az erastin és az RSL3 (47, 48) ferroptózist indukáló szereknek. Ez a hiba hozzájárulhat az S47 egerekben megfigyelt tumorra hajlamos fenotípushoz.,
mutáns p53 érzékenyíti a tumorsejteket Ferroptózisra
vad típusú p53 negatívan szabályozza az SLC7A11 cisztinimportőr expresszióját, amely gátolja a ferroptózisra való érzékenységet (5). Bár ez a Szabályozás normális sejtekben fordul elő, a tumorsejtekben az SLC7A11 egyéb mediátorai dominálnak e gén szabályozásában. Például az Nrf2 mester antioxidáns transzkripciós faktor szabályozhatja az SLC7A11 transzkripciós szintű expresszióját is, az NRF2 pedig kulcsfontosságú szerepet játszik a rákos sejtek ferroptózis elleni védelmében., Például az Nrf2 gátlása a hepatocelluláris rákos sejtekben növeli az erasztin és a szorafenib rákellenes aktivitását in vivo (56). A p53 mutáns formái gátolhatják az NRF2 funkciót közvetlen kölcsönhatással, és egy csoport azt találta, hogy a mutáns p53-mal rendelkező daganatok nagyon alacsony SLC7A11 szintet tartalmaznak, így fokozott érzékenységet mutatnak a ferroptózisra. Nevezetesen, az SLC7A11 túlzott expressziója a mutáns p53 modellekben gyógyszerrezisztenciához vezetett, ami arra utal, hogy az SLC7A11 expresszió szintjét figyelembe kell venni a mutáns p53-vezérelt rákok ferroptózist indukáló vegyületekkel történő célzásakor (57)., Támogatja ezt a feltevést, legutóbbi munkája a colorectalis (CRC) a rák, ahol a mutáció vagy törlését p53 gyakori esemény, kimutatta, hogy az emberi CRC sejtvonalak menedéket mutáns p53 voltak, sokkal érzékenyebb, hogy Erastin-sejt mediált a halál, ha összehasonlítjuk a CRC sejtek WT p53. Ezen eredmények érvényesítése érdekében kimutatták, hogy a p53 hotspot mutáció kopogása mind a HCT116, mind az SW48 sejtekben visszaállította az erastin érzékenységét (51). Ezek az adatok kiemelnek egy új mechanizmust, amellyel a mutáns p53 által vezérelt rákokat célzott terápiával lehet kihasználni.,
következtetés
a p53 szerepe az anyagcserében teljesen egyértelmű, sőt talán intuitív módon is nyilvánvaló: a WT p53 korlátozza a glükóz metabolizmusát és a lipidszintézist, míg a mutáns p53 az ellenkezőjét teszi. A metabolikus szerepének hozzájárulása a p53-as tumorszuppresszióhoz, valamint a mutáns p53-nak a tumor progressziójának vezetéséhez, továbbra is egyértelműen bizonyítottnak kell lennie. A p53 szerepe a ferroptózis szabályozásában, valamint ennek a funkciónak a hozzájárulása a tumorszuppresszióhoz még kevésbé egyértelmű., Míg meggyőző adatokat egér modell támogatja a feltevést, hogy a p53 szabályozza az érzékenységet, a sejtek ferroptosis, ez korlátozható a képesség, a bazális p53, hogy elnyomja a spontán tumor fejlődését, valamint az onkogén-hangsúlyozta egér modellek, egyértelmű, hogy senescence, valamint apoptózis játssza a meghatározó szerepet. Hasonlóképpen, a p53 szabályozhatja a ferroptózis érzékenységét sejttípus-specifikus módon. További vizsgálatok az állati modellek, a figyelmet a ferroptosis különböző szövetekben, meg kell tenni, hogy jobban megértsék a szerepét p53 ferroptosis és ferroptosis tumorszuppresszió., Ezenkívül világosabb képet kell alkotni arról, hogy a p53-cél gének milyen szerepet játszanak a ferroptózis érzékenységében. Ezeknek a kérdéseknek a megoldására nagy szükség van új utakat kell biztosítani a mutáns p53 tumorok elleni küzdelemhez.
szerzői hozzájárulások
KG, SB, TB, AB-K, C-PK, és MM mindegyik e cikk egy-két bekezdését írta. KG és SB tette ezt a számot. KG és MM vázolta a fejezetet.,
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
a bíráló oaf és a kezelő szerkesztő közös hovatartozásukat nyilvánította.
a kiadványban közölt kutatásokat a National Institutes of Health támogatta CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) és T32 CA009171 (TB)., A tartalom kizárólag a szerzők felelőssége, nem feltétlenül képviseli a Nemzeti Egészségügyi Intézetek hivatalos véleményét.
3. Humpton TJ, Vousden KH. A sejtek metabolizmusának és hipoxiájának szabályozása p53-mal. Hideg Tavaszi Harb Perspect Med (2016) 6(7):211-30. doi: 10.1101 / cshperspect.a026146
CrossRef teljes szöveg | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., A p53 tumorszuppresszor negatívan szabályozza a hipoxia által stimulált glikolízist a cél RRAD-on keresztül. Oncotarget (2014) 5(14):5535-46. doi: 10.18632 / oncotarget.2137
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
17. Lee M, Yoon JH. Metabolikus kölcsönhatás a glikolízis és a mitokondriális oxidáció között: a fordított Warburg-hatás és annak terápiás hatása. Világ J Biol Chem (2015) 6(3):148-61. doi:10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., Kimerülése az új p53-cél gén karnitin palmitoiltranszferáz 1C késlelteti a tumor növekedését a neurofibromatózis I. típusú tumor modell. A Sejthalál Különbözik (2013) 20(4):659-68. doi: 10.1038 / cdd.2012.168
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. A szterol-válasz-kötő fehérjék és a prosztatarák downstream effektorainak diszregulációja az androgén függetlenné válás során., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. kopogás-in egerek kiméra humán/murine p53 gén normálisan fejlődnek, és azt mutatják, vad típusú p53 válaszok DNS károsító szerek: egy új orvosbiológiai kutatási eszköz. Onkogén (2001) 20(3):320-8. doi:10.1038 / sj.onc.1204080
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
57. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. A rendszer XC-/glutation tengelyének gátlása szelektíven célozza meg a rákos megbetegedéseket mutáns-p53 felhalmozódással., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















