Genom Spot (Magyar)
A Pathway analysis ilyen gyakori eljárássá vált a bioinformatikában, különösen a génexpresszió tanulmányozásában. Ha megnézzük egy felmérés a legutóbbi papírok úgy tűnik, hogy van egy csomó módon, hogy meg lehet csinálni. Ebben a bejegyzésben, fogom megvitatni a különbségeket két általánosan használt eszközök; DAVID és GSEA.

a két algoritmus mögötti fogalmak nagyon különbözőek., DAVID meghatározza az átfedéseket a felhasználó által megadott génlisták és a kurált adatbázisok között, olyan átfedéseket keresve, amelyek nagyobbak, mint a véletlenszerű véletlen által elvárt. Javíthatja az algoritmus pontosságát olyan háttérfájl biztosításával, amely tartalmazza a kísérletben figyelembe vett/detektált összes gént. A felhasználó által szállított génkészleteket általában olyan gének kiválasztásával generálják, amelyek átadják a szignifikancia küszöböt. A Dávid-eljárás hasonló a többi elérhetőhez, mint például a találékonyság, az AmiGO és a GeneGO., A szignifikancia értékek kiválasztása nagyrészt önkényes, de gyakori, hogy a küszöbértéket az FDR korrigált p<0.05 értékre állítjuk. A küszöbérték módosítása jelentősen megváltoztatja a dúsítási eredményeket. Az alkalmazott statisztikai tesztek közé tartozik a hipergeometria, a Fisher ‘ s exact és a Chi-squared.
a fenti módszerekkel ellentétben, amelyek tetszőleges küszöbértékeket meghaladó gének listáját igénylik, a GSEA olyan eszköz, amely statisztikai algoritmusában minden adatpontot használ. A” klasszikus ” módszerben a géneket a leginkább szabályozott, leginkább szabályozott módon rangsorolják., Maga a rangmutató változó, de két érvényes módszer az aláírt p-érték használata, vagy a hajtásváltozás 90% – os konfidencia intervallumának csökkentése. A vizsgálat alapja annak felmérése, hogy egy génkészlet tagjai a profil egyik végén dúsítottnak tűnnek-e. A dúsítás teszteléséhez a GSEA elvégzi a profil permutációit, kiszámítva a gén dúsítását ezer vagy több alkalommal, hogy empirikusan megbecsülje a p-értékeket. A teljes profilt használó egyéb eszközök analitikai teszteket végeznek, mint például a Wilcoxon és a Kolmogorov-Smirnov teszt.,
tehát most, hogy megértjük az algoritmusok fő különbségeit, elkezdhetjük összehasonlítani a két technika eredményeit. Áttekintettem egy korábban tárgyalt adatkészletet, azacitidin-kezelést az RNS-seq által profilozott AML3 sejtekben. A DESeq által generált differenciális RNS-seq expressziós adatokat fogom kiindulópontként használni DAVID és GSEA összehasonlításához.
DAVID számára készítettem egy háttérfájlt, amely mind a 19 030 génből áll, amelyeket a kísérletben észleltek az észlelési küszöb felett. A jelentős (FDR<0.,05) Az up (2502 gén) és down (2907 gén) irányban található génlistákat külön-külön a DAVID 6.7-nek nyújtottuk be elemzésre, csak a KEGG útvonalakra összpontosítva.
A GSEA esetében ugyanazt a rang metrikus eljárást használtam, amelyet korábban leírtunk. A klasszikus GSEA algoritmust használtam a gsea2-2.2.2-vel.jar végrehajtható, a KEGG génkészletek msigdb v5.1.
az alábbi grafikonon a statisztikailag szignifikáns génkészletek (FDR<0, 05) átfedését külön-külön határoztam meg a felfelé és lefelé szabályozott útvonalakra., A Dávid-eredmények metszéspontját is tartalmaztam, amikor nincs háttér (csak kíváncsiság, nem ajánlott eljárás).
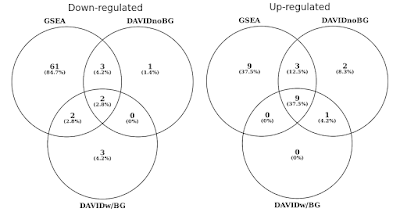
DAVID vs GSEA ugyanazzal a génexpressziós profillal. A számok jelentősen dúsított génkészleteket képviselnek (FDR<0, 05).
észre fogod venni, hogy mind a felfelé, mind lefelé szabályozott irányban a GSEA jelentősebb eredményeket azonosít Dávidhoz képest. Úgy találtam, hogy ez a helyzet más génkészletekben is, mint például a REACTOME and GO., A különbség az érzékenység a két eszköz csapódik le, hogy a különböző algoritmusok, DAVID egy diszkrét gének listáját, versus GSEA, amely minden adatpont. A GSEA sokkal érzékenyebb lesz azon génkészletek azonosításában, amelyeknek sok tagja van, amelyek finom expressziós változásokon mennek keresztül. Egyénileg ezek a változások nem statisztikailag szignifikánsak,de amikor a gének nagy része egy halmazban együttesen az expresszió eltolódása, ez a tendencia statisztikailag nagyon jelentős lehet.,
az érzékenyebb algoritmus mellett DAVID génkészletei és génjegyzetei valóban elavultak. DAVID számos adatforrásának utolsó frissítése 2009 közepétől későig tart. Mint ilyen, az Ensembl számos géncsatlakozása nem támogatható. Valóban, a háttérben fájlt tartalmazott 19,030 csak 13,488 voltak által elismert, DAVID. A frissített génkészletek hiánya szintén aggodalomra ad okot, csakúgy, mint az egyre növekvő számú idézet, amelyet DAVID még mindig kap.,
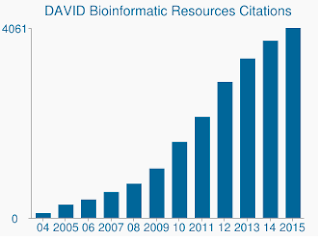
DAVID még mindig évente több ezer idézetet kap, annak ellenére, hogy az alapul szolgáló adatok szomorúan elavultak.
összefoglalva, azt javaslom a kutatóknak, hogy olyan eszközöket használjanak, amelyek egy teljes profilt néznek meg a dúsítás (GSEA, CAMERA, WilcoxGST, Pathifier) meghatározására, ahelyett, hogy génkészleteket küldenének az átfedési elemzéshez. David használata lehet a gyors és egyszerű megoldás, de ez az érzékenység jelentős csökkenésének rovására megy.
további olvasmányok
JA Timmons, KJ Szkop, IJ Gallagher., A torzítás több forrása összezavarja a globális omics-adatok funkcionális dúsításának elemzését. Genombiológia, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. A tudás felhalmozódásának hatása az út gazdagodásának elemzésére. bioRxiv. doi: http://dx.doi.org/10.1101/049288
szerkesztés: 2016 májusában DAVID frissült!
https://david.ncifcrf.gov/content.jsp?file=release.html















