Frontiers in Endocrinology (Italiano)
Introduzione
Il gene soppressore tumorale TP53 è stato il gene umano più studiato dalla sua scoperta quasi 40 anni fa (1). La ragione principale dietro questo stato è il ruolo critico p53 gioca nel prevenire lo sviluppo del cancro, ed è ampiamente considerato come il ” guardiano del genoma.,”Per qualche tempo si è generalmente creduto che il ruolo di p53 nella soppressione del tumore sia in virtù della sua capacità di indurre l’apoptosi, l’arresto del ciclo cellulare e la senescenza delle cellule pre-cancerose (2). Tuttavia, ora è sempre più chiaro che p53 regola molti altri percorsi nella cellula e che questi altri percorsi svolgono anche un ruolo nella capacità di p53 di funzionare come soppressore del tumore (3). In particolare, il ruolo di p53 nella regolazione dei geni coinvolti nel metabolismo e nella ferroptosi è stato implicato nella sua capacità di sopprimere lo sviluppo del tumore., La ferroptosi è una nuova via di morte cellulare caratterizzata per la prima volta nel 2012 e può essere meglio descritta come una forma di morte cellulare dipendente dal ferro e indipendente dalla caspasi determinata dalla formazione di perossidazione lipidica (4). In particolare, due modelli murini contenenti mutazioni ingegnerizzate in p53 che eliminano la capacità di p53 di indurre apoptosi e senescenza mantengono entrambi la capacità di sopprimere lo sviluppo spontaneo del tumore; entrambi questi mutanti mantengono la capacità di transattivare i geni nel metabolismo e nella ferroptosi (5, 6)., Un riassunto dei dati che implicano p53 nella regolazione del metabolismo e della ferroptosi è dettagliato di seguito.
Wild-Type (WT) p53 Regola Positivamente la Fosforilazione Ossidativa e Sopprime il Metabolismo del Glucosio
il Selvaggio-tipo p53 regola la versatilità metabolica delle cellule, favorendo la respirazione mitocondriale oltre la glicolisi, in parte tramite la transattivazione di SCO2 (citocromo c ossidasi assemblea), che svolge un ruolo diretto nella fosforilazione ossidativa (7)., p53 regola anche direttamente la transattivazione di GLS2( Glutaminasi 2); questo enzima consente l’utilizzo di glutammina come fonte di energia per i mitocondri (8). Inoltre, WT p53 regola negativamente la glicolisi reprimendo trascrizionalmente i trasportatori di glucosio GLUT1 e GLUT4 e transattivando RRAD e TIGAR; entrambi sono inibitori della glicolisi (9-11). Infine, p53 lega direttamente e inibisce l’enzima glucosio-6-fosfato deidrogenasi, sopprimendo così il metabolismo del glucosio (12)., È chiaro da questi e da altri studi che negli organismi normali e non stressati, p53 regola direttamente lo stato metabolico in una cellula (Figura 1). Non sorprende che questo gene e molti dei suoi regolatori siano implicati in malattie metaboliche, tra cui obesità e diabete (13).
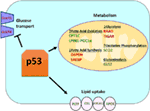
Figura 1. Il ruolo di wild-type (WT) p53 nel metabolismo. I geni regolati positivamente da p53 sono mostrati in verde e i geni regolati negativamente da p53 sono mostrati in rosso., p53 inibisce il trasporto del glucosio, la glicolisi e la sintesi degli acidi grassi mentre promuove l’assorbimento lipidico, l’ossidazione degli acidi grassi, la fosforilazione ossidativa e la glutaminolisi.
Mutante p53 Regola Positivamente Warburg Metabolismo (Glicolisi Aerobica)
In contrasto con la funzione di p53 WT, mutante p53 nelle cellule tumorali e favorisce la glicolisi aerobica, in parte, aumentando il traffico del trasportatore di glucosio GLUT1 alla membrana del plasma, con conseguente aumento del glucosio di importazione (14, 15)., A seguito della mutazione di p53, i livelli ridotti di SCO2 e GLS2 e l’aumento dei livelli di GLUT1 e GLUT4 favoriscono la glicolisi aerobica rispetto alla fosforilazione ossidativa. In questo modo, si ritiene che il mutante p53 contribuisca alla propensione delle cellule tumorali a utilizzare la glicolisi aerobica a favore della fosforilazione ossidativa, o il cosiddetto metabolismo di Warburg (15). Uno dei tratti distintivi del cancro è il metabolismo deregolamentato, generalmente dimostrato da questo passaggio dalla glicolisi aerobica alla fosforilazione ossidativa., Sebbene ciò si traduca in una resa di ATP inferiore e meno efficiente, si ritiene che le cellule tumorali traggano beneficio deviando gli intermedi glicolitici verso percorsi biosintetici necessari per una rapida divisione cellulare (16). Questo interruttore metabolico porta anche ad una diminuzione dell’apoptosi mediata dai mitocondri e ad una segnalazione più efficiente attraverso i metaboliti disponibili nelle cellule tumorali (17).
Una variante genetica comune in TP53 influenza la sua funzione nel metabolismo
C’è un polimorfismo della regione codificante comune di p53 al codone 72, che codifica per prolina (P72) o arginina (R72)., Questa variazione aminoacidica può avere un impatto sulla funzione p53 per quanto riguarda il destino cellulare dopo lo stress. In risposta al danno al DNA, la variante P72 di p53 innesca prevalentemente l’arresto del ciclo cellulare, mentre la variante R72 induce prevalentemente la morte cellulare o l’apoptosi (18, 19). Nonostante queste differenze di funzione, la variazione del codone 72 non è stata costantemente associata alla suscettibilità al cancro (20). Al contrario, negli studi sull’uomo questo polimorfismo è significativamente associato ad un aumento dell’indice di massa corporea e del rischio di diabete (21, 22)., Questa premessa è supportata da studi sui topi, in cui un modello murino per queste varianti codon 72 mostra un aumento del diabete indotto da dieta ad alto contenuto di grassi nei topi con la variante R72, rispetto a P72. In questi studi, i geni bersaglio p53 TNFa e NPC1L1 sono stati identificati come regolatori critici nell’aumento dell’obesità indotta dalla dieta nei topi R72 (23). È interessante notare che la variante R72 ha anche dimostrato di conferire una maggiore sopravvivenza delle cellule in risposta alla privazione dei nutrienti (24)., Questi risultati hanno portato all’ipotesi che la variante R72 di p53 sia sorta ed è stata selezionata per la migrazione delle popolazioni verso nord, dove il freddo richiederebbe un maggiore accumulo di grasso, ma dove la sopravvivenza in risposta alla privazione dei nutrienti sarebbe anche sotto selezione (24).
p53 regola il metabolismo lipidico
Sebbene p53 sia ben noto per la regolazione della glicolisi e del ciclo dell’acido citrico, p53 ha anche dimostrato di svolgere un ruolo nella regolazione del metabolismo lipidico (25)., Si ritiene che WT p53 aumenti l’ossidazione degli acidi grassi inibendo la sintesi degli acidi grassi, agendo così come un regolatore negativo della sintesi lipidica (25). Esistono diversi geni bersaglio p53 con ruoli nel metabolismo lipidico. Sanchez-Macedo e colleghi hanno dimostrato che la carnitina palmitoiltransferasi 1C (CPT1C) è regolata trascrizionalmente da p53; questo enzima aiuta nel trasporto di acidi grassi attivati ai mitocondri., A sostegno di un ruolo per questo gene regolato da p53 nel cancro, questo gruppo ha dimostrato che i topi con deficit di Cpt1c mostrano un ritardo nello sviluppo del tumore e tassi di sopravvivenza più elevati (26). Lipina 1 (LPIN1) è un altro gene bersaglio p53; LPIN1 è necessario per il corretto sviluppo degli adipociti ed è indotto in condizioni di bassi nutrienti (27). Finck e colleghi hanno dimostrato che LPIN1 interagisce con PGC-1α, un altro gene bersaglio p53 noto con un ruolo nel metabolismo, e che questa interazione attiva l’espressione di geni coinvolti nella promozione dell’ossidazione degli acidi grassi (28).,
Oltre a regolare direttamente la trascrizione dei geni coinvolti nel metabolismo lipidico, p53 può anche regolare il metabolismo lipidico in un modo che coinvolge l’interazione diretta proteina–proteina. Ad esempio, la glucosio-6-fosfato deidrogenasi, che è l’enzima limitatore di velocità nella via del pentoso fosfato, si lega ed è direttamente inibita da p53, con conseguente diminuzione della produzione di NADPH e di conseguenza diminuzione della sintesi degli acidi grassi (12)., La famiglia di fattori di trascrizione sterol regulatory element-binding proteins (SREBP) modula l’espressione dei geni coinvolti nella sintesi di colesterolo, acidi grassi, triacilglicerolo e fosfolipidi (29-31). WT p53 reprime la funzione SREBP (32), mentre le forme mutanti di p53 si legano direttamente a SREBP e migliorano la loro funzione trascrizionale, portando ad un aumento dell’attività SREBP nei tumori umani (33, 34). Di conseguenza, il mutante p53 è correlato con una maggiore espressione dei geni della biosintesi degli steroli nei tumori della mammella umana (34, 35)., Infine, AMP – activated protein chinasi (AMPK) è un enzima che viene attivato sotto bassi livelli di nutrienti o stress energetico ed è noto per inibire la sintesi degli acidi grassi interagendo con acetil-CoA-carbossilasi e SREBP-1 (36, 37). Zhou e colleghi hanno dimostrato che il mutante p53 si lega preferenzialmente e inibisce l’AMPK, portando ad un aumento della sintesi degli acidi grassi. Di conseguenza, le proteine mutanti p53 portano ad un aumento della segnalazione AMPK, contribuendo alla crescita cellulare invasiva delle cellule tumorali (33). Un’area meno esplorata è il ruolo di p53 nel trasporto dei lipidi., È stato dimostrato che p53 regola trascrizionalmente l’apolipoproteina B (apoB) e il complesso enzimatico di modifica dell’apoB 1, indicando il ruolo di p53 nella regolazione delle lipoproteine aterogene (38). L’analisi Microarray delle cellule derivate dal fegato umano ha identificato la proteina di trasferimento dei fosfolipidi, la cassetta legante ATP A12 e la lipasi dell’estere carbossilico come tre geni bersaglio p53 che svolgono tutti un ruolo nel trasporto dei lipidi (39, 40)., Nel complesso, anche se è chiaro che p53 svolge un ruolo chiave nella mediazione della sintesi lipidica e del metabolismo, il contributo di questa via, e di questi geni bersaglio p53, alla soppressione del tumore da parte di p53 rimane da determinare (Figura 1).
La ferroptosi è una nuova via di morte cellulare guidata dalla perossidazione lipidica
Nel 2012, Dixon e colleghi hanno scoperto una nuova forma di morte cellulare regolata chiamata ferroptosi. La ferroptosi è una forma di morte cellulare dipendente dal ferro e indipendente dalla caspasi derivante dall’accumulo di lipidi ossidati (4, 41)., Questo processo è guidato dall’inattivazione della glutatione perossidasi 4 (GPX4), un enzima responsabile della conversione degli idroperossidi lipidici letali in alcoli lipidici non tossici, che richiede glutatione per funzionare (41). Si ritiene che la perossidazione degli acidi grassi polinsaturi (PUFA) sia l’impulso trainante per la morte cellulare da parte della ferroptosi. I PUFA contengono protoni bis-allilici che possono essere facilmente astratti e producono radicali che reagiranno con l’ossigeno, creando più radicali e causando una reazione a catena di specie reattive dell’ossigeno lipidico (42)., L’esatto meccanismo di morte cellulare per ferroptosi rimane sconosciuto, ma un’ipotesi è che il danno lipidico porti alla distruzione della membrana plasmatica (43). È stato ipotizzato che la ferroptosi possa essere un meccanismo di soppressione del tumore che funziona eliminando le cellule prive di nutrienti o esposte a stress o infezioni ambientali.,
Regolazione farmacologica della Ferroptosi
La ferroptosi può essere indotta utilizzando inibitori del sistema xc – come erastina, o analoghi come glutammato e sorafenib, che inibiscono l’importazione di cistina, con conseguente glutatione impoverito e successiva inattivazione di GPX4. In alternativa, la ferroptosi può essere indotta da (1S, 3R)-RSL3 (di seguito denominato RSL3), che si lega direttamente e inibisce GPX4 (4, 5, 42). Buthione sulfoximine, FIN56, FINO2, CCl4 e cisplatino sono altri agenti che hanno dimostrato di indurre ferroptosi nelle cellule., Morte da ferroptosis può essere prevenuta mediante l’omissione di perossidazione lipidica, che può essere realizzato utilizzando antiossidanti lipofili, come ferrostatin-1, liproxstatin-1, o vitamina E. chelanti del Ferro come deferoxamina o cicloprox sono un altro strumento utilizzato per sopprimere ferroptosis riducendo i livelli di ferro. L’esaurimento dei PUFA o l’aggiunta di acidi grassi monoinsaturi ai terreni di coltura cellulare possono anche salvare le cellule dalla ferroptosi (42, 44).,
La ferroptosi è implicata nella soppressione del tumore mediata da p53
Nel 2012, Gu e colleghi hanno sviluppato un modello murino in cui tre residui di lisina normalmente acetilati nel dominio di legame al DNA di p53 sono stati mutati in arginina e quindi non possono essere acetilati; questo topo è indicato come il topo 3KR. In particolare, le cellule del topo 3KR non sono in grado di subire apoptosi p53-dipendente, arresto del ciclo cellulare o senescenza, e infatti il mutante 3KR di p53 non riesce a transattivare la maggior parte dei geni bersaglio p53., È interessante notare che questo modello murino non sviluppa spontaneamente il cancro, il che implica che p53 potrebbe sopprimere lo sviluppo tumorale indipendente dalla senescenza o dall’apoptosi (45). Questo gruppo ha scoperto che la proteina mutante 3KR mantiene la capacità di sottoporsi a ferroptosi e regolare il metabolismo della cistina regolando l’espressione dell’importatore di cistina SLC7A11; ciò ha suggerito che la ferroptosi potrebbe essere una via alla base della soppressione del tumore mediata da p53., Quando wild type e 3KR MEFS sono stati trattati con l’induttore di ferroptosi Erastin, è stata osservata quasi il 50% di morte cellulare mentre p53 null MEFS ha mostrato il 20% di morte cellulare; ciò indica che p53 sensibilizza le cellule alla ferroptosi e anche che altri regolatori chiave svolgono un ruolo nella ferroptosi (5). Successivamente, Gu e colleghi hanno identificato un ulteriore sito di acetilazione alla lisina 98 di p53 e hanno generato un modello murino in cui tutti e quattro i siti di acetilazione sono stati mutati in arginina (4KR)., È interessante notare che il mutante 4KR non è stato in grado di regolare i geni coinvolti nella ferroptosi come SLC7A11 e, a differenza del mutante 3KR, non è stato in grado di sopprimere lo sviluppo del tumore (46). Sebbene attualmente correlativo, questi dati implicano il ruolo di p53 nella ferroptosi nella sua capacità di sopprimere lo sviluppo del tumore.
Nelle cellule non trasformate, p53 regola positivamente la Ferroptosi
Oltre a SLC7A11, sono stati scoperti diversi altri geni target diretti di p53 che svolgono un ruolo nella ferroptosi. Questi includono GLS2, PTGS2 e SAT1., Studi di due gruppi separati supportano il ruolo di GLS2 nella ferroptosi, che è noto per diminuire il glutatione e aumentare i livelli di ROS cellulare. Jiang e colleghi hanno usato inibitori della ferroptosi combinati con inibitori della glutaminolisi per inibire la ferroptosi indotta da Erastina, dimostrando così che la ferroptosi richiede glutaminolisi e GLS2 (47). Murphy e colleghi hanno dimostrato che una variante polimorfica di p53 è stata in grado di indurre l’arresto della crescita e la senescenza sia nelle cellule umane che murine, ma non è riuscita a reprimere SLC7A11 o a transattivare GLS2., Questa variante è stata marcatamente compromessa nell’indurre la ferroptosi e sopprimere lo sviluppo del tumore, implicando nuovamente il ruolo di p53 nella soppressione del tumore mediata dalla ferroptosi (48). Un altro gene bersaglio p53 con un ruolo nella ferroptosi è PTGS2, un gene che codifica per l’enzima cicloossigenasi-2. Stockwell e colleghi hanno mostrato per la prima volta che l’induzione della ferroptosi utilizzando Erastin e RSL3 ha portato alla sovraregolazione delle PTG2 (41). In particolare, PTGS2 non è stato sovraregolato dagli induttori di ferroptosi nelle cellule p53-nulle, suggerendo che questa regolazione è dipendente da p53 (5)., Attualmente, l’upregulation di PTGS2 è ampiamente usato come marcatore di ferroptosi (5, 41).
Un recente studio del gruppo Gu ha mostrato che il gene target p53 SAT1 regola la ferroptosi (49). Gli autori hanno identificato SAT1 come un bersaglio diretto di p53 e hanno dimostrato che il silenziamento di SAT1 ha ridotto la morte cellulare indotta da specie reattive dell’ossigeno nelle cellule con WT p53, ma non ha avuto alcun effetto nelle cellule p53-null. Meccanicamente, questo gruppo ha mostrato che SAT1 aumenta il livello e l’attività dell’arachidonato 15-lipossigenasi, un enzima legante il ferro che ossida i PUFA e aumenta la perossidazione lipidica., In particolare, questo studio ha dimostrato che né p53 né SAT1 da soli sembrano essere sufficienti per indurre ferroptosi. Invece, i dati combinati sono più coerenti con la premessa che p53, in virtù dei geni regolatori che contribuiscono alla ferroptosi, regola la sensibilità delle cellule a questa via, piuttosto che indurre direttamente la ferroptosi. Resta da determinare se p53 regoli altri geni coinvolti nella ferroptosi (Figura 2).
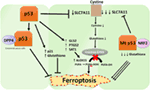
Figura 2. I vari ruoli di p53 nella ferroptosi., L’inibizione della glutatione perossidasi 4 (GPX4), l’enzima chiave che catalizza la conversione degli acidi grassi polinsaturi (PUFA) contenenti perossidi in alcoli, è il fattore chiave della ferroptosi. A seconda del contesto, p53 può sopprimere la ferroptosi (come nelle cellule tumorali del colon-retto) o promuovere la ferroptosi. Il mutante p53 sensibilizza le cellule alla ferroptosi ancor più del selvaggio p53.,
In alcune cellule, p53 regola negativamente la ferroptosi
Uno studio recentemente pubblicato da Tarangelo e colleghi mostra che p53 regola negativamente la ferroptosi nelle cellule tumorali (50). Questo gruppo ha scoperto che il pre-trattamento delle cellule con Nutlin-3, un composto che stabilizza p53 ritarda l’insorgenza della ferroptosi in diversi tipi di cellule. È stato riscontrato che l’insorgenza ritardata della ferroptosi dipende da CDKN1A (codifica p21), un obiettivo trascrizionale p53 critico., Il meccanismo attraverso il quale p21 ritarda la ferroptosi deve ancora essere chiarito, ma si ritiene che la conservazione del glutatione intracellulare possa essere un fattore che contribuisce alla ridotta sensibilità alla ferroptosi. Gli autori concludono che l’asse p53-p21 consente alle cellule tumorali di sopravvivere in condizioni di stress metabolico, come la privazione della cistina, sopprimendo l’insorgenza della ferroptosi (50). Uno studio recente ha dimostrato che p53 inibisce la ferroptosi nelle cellule tumorali del colon-retto legandosi all’enzima dipeptidil-peptidasi-4 (DPP4), che è un modulatore della ferroptosi e del metabolismo lipidico., Meccanicamente, questo studio ha dimostrato che p53 antagonizza la ferroptosi sequestrando DPP4 in un pool inattivo enzimatico nucleare. In assenza di p53, DPP4 è libero di interagire e formare un complesso con NOX1; questo porta ad un aumento della perossidazione lipidica e della ferroptosi. L’inibizione di DPP4 sopprime significativamente la ferroptosi, mentre la sovraespressione di DPP4 innesca la sensibilità all’Erastina, in particolare nelle cellule impoverite di p53 (51). Il controllo bidirezionale della ferroptosi da parte di p53 attraverso meccanismi dipendenti dalla trascrizione e indipendenti dalla trascrizione può essere dipendente dal contesto o dal tipo cellulare (Figura 2).,
Il polimorfismo P47S di TP53 Colpisce la ferroptosi e la soppressione del tumore
Oltre alle mutazioni missense, ci sono diversi polimorfismi a singolo nucleotide (SNPs) funzionalmente significativi nel gene TP53 e altre proteine note per regolare questa via (come MDM2 e MDM4). La variante Pro47Ser (di seguito S47) è il secondo SNP più comune trovato nella regione di codifica p53 (dopo Pro72Arg) che altera la sequenza aminoacidica della proteina., Per chiarire meglio l’impatto di questa variante sulla funzione p53 e sul rischio di cancro, il gruppo Murphy ha generato un modello di topo knock-in p53 umanizzato, in cui gli esoni 4-9 di p53 murino sono stati sostituiti da esoni p53 umani contenenti il tipo wild o la variante S47 (52-55). La maggior parte dei topi S47 ha sviluppato spontaneamente tumori di vari tipi istologici, in particolare il cancro del fegato, tra i 12 ei 18 mesi di età, a differenza dei topi WT p53 (48)., Nei fibroblasti embrionali di topo e nelle linee cellulari linfoblastoidi umane, la variante S47 ha mostrato una compromissione della morte cellulare programmata in risposta al cisplatino e ad altri stress genotossici. Meccanicamente, la variante S47 è difettosa per la transattivazione di geni coinvolti nel metabolismo, come Gls2 (glutaminasi 2) e Sco2 (48). Coerente con il ruolo di Gls2 nella ferroptosi, questo gruppo ha scoperto che le cellule S47 erano marcatamente resistenti agli agenti induttori della ferroptosi Erastin e RSL3 (47, 48). Questo difetto può contribuire al fenotipo tumorale osservato nei topi S47.,
Il mutante p53 sensibilizza le cellule tumorali alla Ferroptosi
Wild-type p53 regola negativamente l’espressione dell’importatore di cistina SLC7A11, che inibisce la sensibilità alla ferroptosi (5). Sebbene questa regolazione si verifichi nelle cellule normali, nelle cellule tumorali, altri mediatori di SLC7A11 sembrano predominare nella regolazione di questo gene. Ad esempio, il master antiossidante fattore di trascrizione NRF2 può anche regolare l’espressione di SLC7A11 a livello trascrizionale, e NRF2 è stato implicato come un giocatore chiave nella protezione delle cellule tumorali contro ferroptosi., Ad esempio, l’inibizione di NRF2 nelle cellule tumorali epatocellulari aumenta l’attività anti-cancro di Erastin e Sorafenib in vivo (56). Le forme mutanti di p53 possono inibire la funzione NRF2 per interazione diretta e un gruppo ha scoperto che i tumori con p53 mutante contengono livelli molto bassi di SLC7A11 e quindi mostrano una maggiore sensibilità alla ferroptosi. In particolare, la sovraespressione di SLC7A11 nei modelli mutanti p53 ha portato alla resistenza ai farmaci, suggerendo che i livelli di espressione di SLC7A11 devono essere considerati quando si prendono di mira i tumori guidati da mutanti p53 con composti induttori della ferroptosi (57)., A sostegno di questa premessa, un recente lavoro sul cancro del colon-retto (CRC), in cui la mutazione o la delezione di p53 è un evento frequente, ha dimostrato che le linee cellulari CRC umane che ospitano il mutante p53 erano molto più sensibili alla morte cellulare mediata da Erastina rispetto alle cellule CRC con WT p53. Per convalidare questi risultati, hanno dimostrato che il knock in di una mutazione hotspot p53 in entrambe le cellule HCT116 e SW48 ha ripristinato la sensibilità all’Erastina (51). Questi dati evidenziano un nuovo meccanismo attraverso il quale i tumori guidati dal mutante p53 possono essere sfruttati utilizzando una terapia mirata.,
Conclusione
Il ruolo di p53 nel metabolismo è abbastanza chiaro e forse anche intuitivamente ovvio: WT p53 limita il metabolismo del glucosio e la sintesi lipidica, mentre il mutante p53 sembra fare il contrario. Il contributo del suo ruolo metabolico alla soppressione del tumore da parte di p53 e alla capacità del mutante p53 di guidare la progressione del tumore, rimane da dimostrare inequivocabilmente. Il ruolo di p53 nella regolazione della ferroptosi e il contributo di questa funzione alla soppressione del tumore è ancora meno chiaro., Mentre i dati convincenti dai modelli del topo sostengono la premessa che p53 regola la sensibilità delle cellule a ferroptosis, questo può essere limitato alla capacità di p53 basale di sopprimere lo sviluppo spontaneo del tumore e nei modelli del topo oncogene-sollecitati, è chiaro che la senescenza e l’apoptosi svolgono il ruolo predominante. Allo stesso modo, p53 può regolare la sensibilità della ferroptosi in modo specifico per il tipo di cellula. Ulteriori studi su modelli animali, con attenzione alla ferroptosi in diversi tessuti, devono essere fatti per comprendere meglio il ruolo di p53 nella ferroptosi e nella ferroptosi nella soppressione del tumore., Inoltre, è necessario ottenere un’idea più chiara di quali geni bersaglio p53 svolgono un ruolo nella sensibilità alla ferroptosi. La risoluzione di queste domande dovrebbe prevedere nuove strade tanto necessarie per combattere i tumori con p53 mutante.
Contributi dell’autore
KG, SB, TB, AB-K, C-PK e MM hanno scritto ciascuno da uno a due paragrafi di questo articolo. KG e SB hanno fatto la figura. KG e MM hanno delineato il capitolo.,
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Il revisore OAF e l’editor di gestione hanno dichiarato la loro affiliazione condivisa.
Riconoscimenti
La ricerca riportata in questa pubblicazione è stata supportata dal National Institutes of Health con i numeri di premio CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK) e T32 CA009171 (TB)., Il contenuto è di esclusiva responsabilità degli autori e non rappresenta necessariamente le opinioni ufficiali del National Institutes of Health.
3. Humpton TJ, Vousden KH. Regolazione del metabolismo cellulare e ipossia da p53. Primavera fredda Harb Perspect Med (2016) 6(7):211-30. doi: 10.1101 / cshperspect.a026146
CrossRef Full Text/Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., Il soppressore tumorale p53 regola negativamente la glicolisi stimolata dall’ipossia attraverso il suo bersaglio RRAD. Oncotarget (2014) 5(14):5535-46. doi:10.18632 / oncotarget.2137
PubMed Abstract | CrossRef Full Text/Google Scholar
17. Lee M, Yoon JH. Interazione metabolica tra glicolisi e ossidazione mitocondriale: l’effetto Warburg inverso e la sua implicazione terapeutica. Mondo J Biol Chem (2015) 6(3):148-61. doi: 10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., L’esaurimento del nuovo gene p53-target carnitina palmitoiltransferasi 1C ritarda la crescita del tumore nel modello di tumore di tipo I neurofibromatosi. La morte cellulare differisce (2013) 20(4):659-68. doi: 10.1038 / cdd.2012.168
PubMed Abstract | CrossRef Full Text/Google Scholar
30. Il nostro sito utilizza cookie tecnici e di terze parti per migliorare la tua esperienza di navigazione. Disregolazione delle proteine leganti gli elementi di risposta degli steroli e degli effettori a valle nel cancro alla prostata durante la progressione verso l’indipendenza dagli androgeni., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. I topi knock-in con un gene p53 umano/murino chimerico si sviluppano normalmente e mostrano risposte p53 di tipo selvaggio agli agenti dannosi del DNA: un nuovo strumento di ricerca biomedica. Oncogene (2001) 20(3):320-8. doi:10.1038 / sj.onc.1204080
PubMed Abstract | CrossRef Full Text/Google Scholar
57. Il nostro sito utilizza cookie tecnici e di terze parti. Inibendo il sistema l’asse XC – / glutatione mira selettivamente ai tumori con accumulo di mutante-p53., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















