Genome Spot (Italiano)
L’analisi della via è diventata una procedura comune in bioinformatica, specialmente nello studio dell’espressione genica. Se si guarda a un sondaggio di documenti recenti sembra che ci sia un sacco di modi che può essere fatto. In questo post, discuterò le differenze tra due strumenti comunemente usati; DAVID e GSEA.

I concetti alla base dei due algoritmi sono molto diversi., DAVID determina sovrapposizioni tra gli elenchi di geni forniti dall’utente e i database curati, alla ricerca di sovrapposizioni più grandi di quelle previste per caso. È possibile migliorare la precisione dell’algoritmo fornendo un file di sfondo che contiene tutti i geni che sono stati considerati/rilevati nell’esperimento. I set di geni forniti dall’utente sono normalmente generati selezionando geni che superano una soglia di significatività. La procedura DAVID è simile ad altri disponibili come Ingegno, AmiGO e GeneGO., La selezione dei valori di significatività è in gran parte arbitraria, ma è comune impostare la soglia su FDR adjusted p<0.05. La modifica della soglia altererà notevolmente i risultati dell’arricchimento. I test statistici impiegati includono ipergeometrico, esatto di Fisher e Chi-quadrato.
In contrasto con i metodi di cui sopra che richiedono elenchi di geni che superano soglie arbitrarie, GSEA è uno strumento che utilizza ogni datapoint nel suo algoritmo statistico. Nel metodo” classico ” i geni sono classificati da più up-regulated a più down-regulated., La metrica rank stessa varia, ma due metodi validi devono utilizzare il valore p firmato o un intervallo di confidenza inferiore al 90% della modifica della piega. La base del test è valutare se i membri di un set genetico appaiono arricchiti ad un’estremità del profilo. Per testare l’arricchimento, GSEA esegue permutazioni del profilo, calcolando l’arricchimento del gene impostato mille o più volte per stimare i valori p empiricamente. Altri strumenti che utilizzano l’intero profilo eseguono test analitici come il test Wilcoxon e Kolmogorov-Smirnov.,
Quindi, ora che comprendiamo le principali differenze negli algoritmi, possiamo iniziare a confrontare i risultati di queste due tecniche. Ho rivisitato un set di dati che ho discusso prima, il trattamento con azacitidina in cellule AML3 profilate da RNA-seq. Userò i dati di espressione differenziale RNA-seq generati da DESeq come punto di partenza per confrontare DAVID e GSEA.
Per DAVID, ho fatto un file di sfondo composto da tutti i geni 19,030 rilevati nell’esperimento sopra la soglia di rilevamento. Il significativo (FDR<0.,05) gli elenchi di geni nella direzione up (2502 geni) e down (2907 geni) sono stati presentati separatamente a DAVID 6.7 per l’analisi, concentrandosi solo sui percorsi di KEGG.
Per GSEA, ho usato la stessa procedura metrica di rango descritta in precedenza. Ho usato il classico algoritmo GSEA con il gsea2-2.2.2.eseguibile jar, utilizzando set di geni KEGG da MSigDB v5.1.
Nel grafico sottostante, ho determinato la sovrapposizione di set genetici statisticamente significativi (FDR< 0.05) separatamente per percorsi regolati su e giù., Ho anche incluso un’intersezione dei risultati di DAVID quando non viene fornito alcun background (solo per curiosità, non una procedura raccomandata).
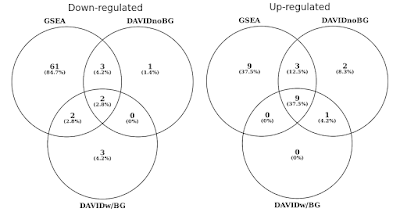
DAVID vs GSEA utilizzando lo stesso profilo di espressione genica. I numeri rappresentano insiemi di geni che sono significativamente arricchiti (FDR<0.05).
Noterai che sia nella direzione regolata su e giù, GSEA identifica risultati più significativi rispetto a DAVID. Ho scoperto che questo era anche il caso in altre librerie di set di geni come REACTOME e GO., La differenza nella sensibilità tra i due strumenti si riduce ai diversi algoritmi, con DAVID che utilizza un elenco discreto di geni, rispetto a GSEA che utilizza ogni punto dati. GSEA sarà molto più sensibile nell’identificare insiemi di geni che hanno molti membri che stanno subendo sottili cambiamenti nell’espressione. Individualmente, questi cambiamenti non sono statisticamente significativi, ma quando una grande frazione di geni in un insieme collettivamente spostamento di espressione, tale tendenza potrebbe essere molto significativo statisticamente parlando.,
Oltre all’algoritmo più sensibile, i set di geni e le annotazioni geniche di DAVID sono davvero obsoleti. L’ultimo aggiornamento delle molte fonti di dati di DAVID è a metà-fine 2009. Come tale, molte adesioni gene da Ensembl potrebbero non essere supportati. Infatti, dal mio file di sfondo che conteneva 19.030 solo 13.488 sono stati riconosciuti da DAVID. Anche la mancanza di set genetici aggiornati è una preoccupazione, così come il numero sempre crescente di citazioni che DAVID riceve ancora.,
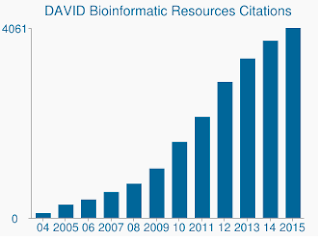
DAVID riceve ancora migliaia di citazioni all’anno nonostante i dati sottostanti siano tristemente obsoleti.
In conclusione, consiglierei ai ricercatori di utilizzare strumenti che esaminano un intero profilo per determinare l’arricchimento (GSEA, CAMERA, WilcoxGST, Pathifier) piuttosto che inviare set di geni per l’analisi della sovrapposizione. Utilizzando DAVID potrebbe essere l’opzione semplice e veloce, ma questo viene a scapito di una massiccia riduzione della sensibilità.
Ulteriori letture
JA Timmons, KJ Szkop, IJ Gallagher., Molteplici fonti di bias confondono l’analisi di arricchimento funzionale dei dati globali-omici. Biologia del genoma, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Impatto dell’accumulo di conoscenze sull’analisi dell’arricchimento del percorso. Biorossiv. doi:http://dx.doi.org/10.1101/049288
MODIFICA: A maggio 2016, DAVID è stato aggiornato!
https://david.ncifcrf.gov/content.jsp?file=release.html















