内分泌学におけるFrontiers
はじめに
腫瘍サプレッサー遺伝子TP53は、40年近く前に発見されて以来、最も研究されてきたヒト遺伝子である(1)。 この状態の背後にある主な理由は、p53が癌の発症を予防する上で重要な役割を果たしていることであり、広く”ゲノムの保護者”とみなされています。,”しばらくの間、腫瘍抑制におけるp53の役割は、前癌性細胞のアポトーシス、細胞周期停止、および老化を誘導する能力によるものであると一般に信じられていた(2)。 しかしながら、p53が細胞内の他の多くの経路を調節し、これらの他の経路もまた、p53が腫瘍抑制剤として機能する能力において役割を果たすことがますます明らかになっている(3)。 特に、代謝およびフェロプトーシスに関与する遺伝子の調節におけるp53の役割は、腫瘍の発生を抑制する能力に関与している。, Ferroptosisは、最初の2012年に特徴付けられる新規細胞死経路であり、最高の脂質過酸化(の形成によって駆動される細胞死の鉄依存性、カスパーゼ非依存型として記述することができます4)。 具体的には、p53のアポトーシスと老化を誘導するp53の能力を排除するp53の設計変異を含む二つのマウスモデルは、両方の自発的な腫瘍の開発を抑制する能力を保持し、これらの変異体の両方は、代謝およびferroptosis(5、6)に遺伝子をトランス活性化する能力を保持します。, 代謝およびフェロプトーシスの調節におけるp53を関与させるデータの要約を以下に詳述する。
野生型(WT)p53は積極的に酸化的リン酸化を調節し、グルコース代謝を抑制する
野生型p53は、酸化的リン酸化に直接の役割を果たしているSCO2(シトクロムcオキシダーゼアセンブリ)のトランスアクティブ化を介して、解糖よりもミトコンドリア呼吸を支持することによって、細胞の代謝の多様性を調節する(7)。, p53はまた直接GLS2(グルタミナーゼ2)のtransactivationを調整します;この酵素はmitochondria(8)のためのエネルギー源としてグルタミンの使用法を可能にします。 さらに、WT p53負転写グルコーストランスポーター GLUT1とGLUT4を抑制することによって、およびrradとTIGARをトランスアクティブ化することによって解糖を調節する;どちらも解糖の阻害剤である(9-11)。 最後に、p53はまた、直接結合し、このようにグルコース代謝(抑制、酵素グルコース-6-リン酸デヒドロゲナーゼを阻害します12)。, これらの研究および他の研究から、正常でストレスのない生物では、p53が細胞内の代謝状態を直接調節することは明らかである(図1)。 驚くことではないが、この遺伝子およびその調節因子の多くは、肥満および糖尿病(を含む代謝性疾患に関与している13)。
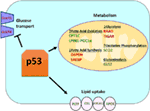
図1. 代謝における野生型(WT)p53の役割。 P53によって正に調節される遺伝子は緑色で示され、p53によって負に調節される遺伝子は赤色で示されている。, p53は脂質の通風管、脂肪酸の酸化、酸化リン酸化およびglutaminolysisを促進する間、ブドウ糖の輸送、解糖および脂肪酸の統合を禁じます。
変異p53は積極的にWarburg代謝(好気性解糖)を調節します
WT p53の機能とは対照的に、腫瘍細胞における変異p53は、部分的には、原形質膜へのグルコーストランスポーター GLUT1の人身売買を強化することによって、好気性解糖を支持します(14、15)。, P53の突然変異に続いて、SCO2およびGLS2の減少レベルおよびGLUT1およびGLUT4の増加レベルは、酸化的リン酸化よりも好気性解糖を支持する。 このようにして、変異p53は、酸化的リン酸化、またはいわゆるWarburg代謝(の賛成で好気性解糖を利用する腫瘍細胞の傾向に寄与すると考えられている15)。 がんの特徴の一つは、一般的に好気性解糖から酸化的リン酸化へのこのスイッチによって示される代謝の規制緩和です。, これはより低く、より少なく有効なATPの収穫で起因するが、癌細胞は急速な細胞分裂に必要な生合成経路に解糖性の中間物を転換することによって寄与すると考えられている(16)。 このスイッチの代謝にもつながる減少したミトコンドリアを介したアポトーシスおよびより効率的なシグナル伝達により代謝物の癌細胞(17).
TP53の共通の遺伝的変異は、代謝におけるその機能に影響を与えます
p53の共通のコード領域多型がありますコドン72で、プロリン(P72)またはアルギニン(R72)のいずれかをコードする。, このアミノ酸の変化は圧力の後で細胞の運命に関してp53機能に影響を与えることができます。 DNA損傷に応答して、P72変異体のP53は主に細胞周期停止を引き起こし、R72変異体は主に細胞死またはアポトーシスを誘導する(18、19)。 機能のこれらの違いにもかかわらず、コドン72の変化は一貫して癌感受性(と関連付けられていない20)。 対照的に、ヒトの研究では、この多型が有意に増加したボディマス指数と糖尿病(リスクに関連付けられている21、22)。, この前提は、これらのコドン72変異体のマウスモデルは、P72と比較して、R72変異体を有するマウスで増加した高脂肪食誘発性糖尿病を示しているマウスでの研究によってサポートされています。 これらの研究では、P53標的遺伝子TNFaおよびNPC1L1は、R72マウス(23)における食事誘発性肥満の増加における重要な調節因子として同定された。 興味深いことに、R72変異体はまた、栄養欠乏に応答して細胞の生存の増加を与えることが示されている(24)。, これらの知見は、p53のR72バリアントが発生し、寒い天候が増加した脂肪蓄積を必要とするが、栄養不足に応答して生存も選択下にあるだろうところ、北に移行した集団としてのために選択されたという仮説につながっている(24)。
p53は脂質代謝を調節する
p53は解糖とクエン酸サイクルを調節することでよく知られていますが、p53は脂質代謝を調節する役割を果たすことが示されています(25)。, WT p53は脂肪酸合成を阻害しながら脂肪酸酸化を促進し、したがって脂質合成の負の調節因子として作用すると考えられている(25)。 脂質代謝における役割を持ついくつかのp53標的遺伝子があります。 今回、Sanchez-Macedoたちの研究グループは、カルニチンパルミトイルトランスフェラーゼ1C(CPT1C)がp53によって転写調節されていることを明らかにし、この酵素が活性化脂肪酸のミトコンドリアへの輸送を助けることを明らかにした。, 癌におけるこのp53調節遺伝子の役割をサポートするために、このグループは、Cpt1c欠損マウスが遅れた腫瘍発達と高い生存率を表示することを示した(26)。 リピン1(LPIN1)は、別のp53標的遺伝子であり、LPIN1は、適切な脂肪細胞の開発のために必要であり、低栄養条件下で誘導される(27)。 今回、Finckたちは、LPIN1がPGC-1αと相互作用し、代謝に関与するp53標的遺伝子として知られており、この相互作用によって脂肪酸酸化の促進に関与する遺伝子の発現が活性化されることを明らかにした(28)。,
脂質代謝に関与する遺伝子の転写を直接調節することに加えて、p53はまた、直接タンパク質–タンパク質相互作用を含む方法で脂質代謝を調節 例えば、ペントースリン酸経路の律速酵素であるグルコース-6-リン酸デヒドロゲナーゼは、p53に結合して直接阻害され、NADPH産生が減少し、結果的に脂肪酸合成が減少する(12)。, 転写因子のステロール調節要素結合タンパク質(SREBP)ファミリーは、コレステロール、脂肪酸、トリアシルグリセロール、およびリン脂質合成(29-31)に関与する遺伝子の発 WT p53はSREBP機能(32)を抑制し、p53の変異型はSREBPに直接結合し、それらの転写機能を増強し、ヒト腫瘍におけるsrebp活性の増加につながる(33、34)。 その結果、変異体p53は、ヒト乳房腫瘍(におけるステロール生合成遺伝子の高発現と相関している34、35)。, 最後に、AMP活性化プロテインキナーゼ(AMPK)は、低栄養レベルまたはエネルギーストレス下で活性化され、アセチル-CoA-カルボキシラーゼおよびSREBP-1(36、37)との相互作用によって脂肪酸合成を阻害することが知られている酵素である。 Zhouたちは、変異体p53がAMPKに優先的に結合して阻害し、脂肪酸合成の増加につながることを実証した。 その結果、変異p53タンパク質は、腫瘍細胞の侵襲性細胞増殖に寄与する、増加AMPKシグナル伝達につながる(33)。 より少ない探索領域は、脂質輸送におけるp53の役割である。, これは、p53が転写アポリポタンパク質B(apoB)とアポブ編集酵素複合体1を調節することが示されており、アテローム形成リポタンパク質(38)を調節する ヒト肝由来細胞のマイクロアレイ解析は、リン脂質転送タンパク質、ATP結合カセットA12、およびカルボキシルエステルリパーゼは、すべての脂質輸送(39、40)において役割を果たしている三つのp53標的遺伝子として同定された。, 全体として、p53が脂質合成と代謝を媒介する重要な役割を果たしていることは明らかであるが、この経路およびこれらのp53標的遺伝子がp53による腫瘍抑制に対する寄与は依然として決定されていない(図1)。
Ferroptosisは脂質過酸化によって駆動される新しい細胞死経路である
2012年、Dixonらはferroptosisと呼ばれる新しい形態の調節された細胞死を発見した。 Ferroptosisは、酸化脂質(の蓄積から生じる細胞死の鉄依存性、カスパーゼ非依存型である4、41)。, このプロセスは、致命的な脂質ヒドロペルオキシドを非毒性脂質アルコールに変換する酵素であるグルタチオンペルオキシダーゼ4(GPX4)の不活性化によって駆動され、機能するためにグルタチオンを必要とする(41)。 多価不飽和脂肪酸(PUFAs)の過酸化は、フェロプトーシスによる細胞死の推進力であると考えられている。 Pufaは、容易に抽出することができ、より多くのラジカルを作成し、脂質反応性酸素種(の連鎖反応をもたらす、酸素と反応するラジカルを生成することができるビス-アリルプロトンを含む42)。, フェロトーシスによる細胞死の正確なメカニズムは不明のままであるが、一つの仮説は、脂質損傷が原形質膜(43)の破壊につながるということです。 フェロトーシスは、栄養素が奪われているか、環境ストレスや感染にさらされている細胞を排除することによって働く腫瘍抑制のメカニズムである,
フェロプトーシスの薬理学的調節
フェロプトーシスは、エラスチンなどのxc系の阻害剤、またはグルタミン酸やソラフェニブなどの類似体を用いて誘導され、シスチンの輸入を阻害し、グルタチオンの枯渇とGPX4のその後の不活性化をもたらす。 あるいは、(1S,3R)-RSL3(以下、RSL3と称する)によってフェロトーシスが誘導され、GPX4(4,5,42)に直接結合し阻害される。 Buthioneのスルホキシミン、FIN56、FINO2、CCl4およびcisplatinは細胞のferroptosisを引き起こすために示された他のエージェントです。, フェロプトーシスによる死は、フェロスタチン-1、リプロキスタチン-1、またはビタミンEなどの親油性抗酸化物質を使用することによって達成することができる脂質過酸化を抑制することによって防止することができる。 Pufaを枯渇させるか、または細胞培養培地に一価不飽和脂肪酸を添加することはまた、フェロプトーシス(から細胞を救出することができる42、44)。,
Ferroptosisはp53を介した腫瘍抑制に関与している
2012年、Guらは、p53のDNA結合ドメイン内の三つの正常にアセチル化されたリジン残基がアルギニンに変異したため、アセチル化することができなかったマウスモデルを開発した。このマウスは3KRマウスと呼ばれている。 特に、3KRマウスからの細胞は、p53依存性のアポトーシス、細胞周期停止、または老化を受けることができず、実際にp53の3KR変異体は、p53標的遺伝子の, 興味深いことに、このマウスモデルは自発的にp53が老化またはアポトーシス(の独立した腫瘍の開発を抑制できることを意味し、癌を開発していない45)。 このグループは、変異3KRタンパク質は、シスチン輸入SLC7A11の発現を調節することによってフェロプトーシスを受け、シスチン代謝を調節する能力を保持することを発見した;これはフェロプトーシスは、p53を介した腫瘍抑制の根底にある一つの経路である可能性があることを示唆した。, 野生型と3KR Mefは、フェロプトーシス誘導エラスチンで処理したとき、ほぼ50%の細胞死が観察されたp53ヌルMefは20%の細胞死を示したのに対し、これはp53がフェロプトーシスに細胞を感作することを示し、また、他の重要なレギュレータはまた、フェロプトーシスにおいて役割を果たしていることを示している(5)。 その後、Guたちは、リジン98のp53に追加のアセチル化部位を同定し、四つのアセチル化部位をすべてアルギニン(4KR)に変異させたマウスモデルを作, 興味深いことに、4KR変異体は、SLC7A11のようなferroptosisに関与する遺伝子を調節することができなかった、と3KR変異体とは異なり、腫瘍の発生を抑制することができませんでした(46)。 現時点では相関的ではあるが、これらのデータは、腫瘍の発生を抑制する能力におけるフェロプトーシスにおけるp53の役割を関与させる。
非形質転換細胞では、p53は積極的にフェロプトーシスを調節します
SLC7A11に加えて、いくつかの他の直接p53標的遺伝子は、フェロプトーシスに これらには、GLS2、PTGS2、およびSAT1が含まれます。, 二つの別々のグループからの研究は、グルタチオンを減少させ、細胞のROSレベルを増加させることが知られているferroptosisにおけるGLS2の役割をサポートし Jiangたちは、フェロプトーシス阻害剤とグルタミノリシス阻害剤を組み合わせてエラスチン誘導フェロプトーシスを阻害し、フェロプトーシスにはグルタミノリシスとGLS2が必要であることを示した(47)。 Murphyたちは、p53の多型変異体がヒトおよびマウス細胞の両方で増殖停止および老化を誘導することができたが、SLC7A11を抑制またはGLS2をトランスアクティブ化することができなかったことを明らかにした。, このバリアントは著しくこのように再びフェロプトーシスを介した腫瘍抑制におけるp53の役割を関与し、フェロプトーシスを誘導し、腫瘍の発達を抑制する際に損なわれた(48)。 フェロプトーシスにおける役割を持つ別のp53標的遺伝子はPTGS2であり、これは酵素シクロオキシゲナーゼ-2をコードする遺伝子である。 今回、Stockwellたちは、エラスチンとRSL3を用いたフェロプトーシスの誘導がPTGS2のアップレギュレーションにつながることを最初に示した(41)。 特に、PTGS2は、この規制がp53依存であることを示唆し、p53ヌル細胞におけるフェロプトーシス誘導物質によってアップレギュレートされていなかった(5)。, 現在、PTGS2のアップレギュレーションは広くフェロトーシスマーカー(5、41)として使用されています。
Guグループによる最近の研究では、p53標的遺伝子SAT1がフェロプトーシスを調節することが示された(49)。 著者らは、SAT1をp53の直接標的と同定し、SAT1のサイレンシングはWT p53を有する細胞における活性酸素種によって誘導される細胞死を減少させるが、p53ヌル細胞には影響を及ぼさないことを示した。 機械論的に、このグループはSAT1がarachidonate15lipoxygenase、PUFAsを酸化し、脂質過酸化を高める鉄結合酵素のレベルそして活動を高めることを示しました。, 特に、この研究は、p53もSAT1だけではフェロプトーシスを誘導するのに十分であるように見えることを示した。 代わりに、組み合わせたデータは、p53は、ferroptosisに寄与する遺伝子を調節するおかげで、この経路に対する細胞の感受性を調節するのではなく、直接ferroptosisを誘導するという前提とより一致している。 P53がフェロプトーシスに関与する他の遺伝子を調節するかどうかは、依然として決定されている(図2)。
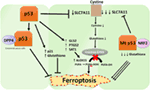
図2. フェロプトーシスにおけるp53の様々な役割。, 過酸化物を含む多価不飽和脂肪酸(Pufa)のアルコールへの変換を触媒する重要な酵素であるグルタチオンペルオキシダーゼ4(GPX4)の阻害は、フェロプトーシスの主要なドライバーである。 文脈によって、p53はferroptosisを抑制できます(結腸直腸癌のセルのような)またはferroptosisを促進して下さい。 変異p53は、野生型p53よりもさらにフェロプトーシスに細胞を感作する。,
いくつかの細胞では、p53は負のFerroptosisを調節します
Tarangeloらによって最近発表された研究は、p53が癌細胞におけるferroptosisを負に調節することを示しています(50)。 このグループはNutlin-3、p53を安定させる混合物とのセルを前処理することが複数の細胞のタイプのferroptosisの手始めを遅らせることを見つけました。 フェロプトーシスの遅延発症は、CDKN1A(エンコードp21)、重要なp53転写標的に依存することがわかった。, P21遅延ferroptosisを介してメカニズムはまだ解明されていないが、細胞内グルタチオンの保存は減少ferroptosis感度のための寄与因子であるかもしれないと考えられている。 著者らは、p53–p21軸は、フェロプトーシスの発症を抑制することによって、シスチン欠乏などの代謝ストレス条件下で癌細胞が生存することを可能にすると結論付けている(50)。 最近の研究では、p53は、フェロプトーシスと脂質代謝の変調器である酵素ジペプチジルペプチダーゼ-4(DPP4)に結合することにより、結腸直腸癌の細胞におけるフェロプトーシスを阻害することを示した。, 機械的に、本研究では、p53は、核酵素不活性プールでDPP4を隔離することによってferroptosisに拮抗することを示した。 P53の非存在下では、DPP4はNOX1と相互作用して複合体を形成することが自由であり、これは脂質過酸化およびフェロプトーシスの増加をもたらす。 DPP4の阻害は、DPP4の過剰発現は、特にp53枯渇した細胞(51)において、エラスチン感受性をトリガするのに対し、大幅にフェロプトーシスを抑制する。 転写依存性および転写非依存性メカニズムによるp53によるフェロプトーシスの双方向制御は、コンテキストまたは細胞型依存性であり得る(図2)。,
TP53のP47S多型は、フェロプトーシスと腫瘍抑制に影響を与えます
ミスセンス変異に加えて、TP53遺伝子およびこの経路を調節することが知られている他のタンパク質(MDM2およびMDM4など)には、いくつかの機能的に重要な一塩基多型(Snp)があります。 Pro47Serバリアント(以下S47)は、タンパク質のアミノ酸配列を変更するp53コード領域(Pro72Argの後)で見つかった第二の最も一般的なSNPです。, より良いp53機能と癌リスクに対するこのバリアントの影響を解明するために、マーフィーグループは、ヒト化p53ノックインマウスモデルを生成し、マウスp53のエクソン4-9は、野生型またはS47変異体(52-55)のいずれかを含むヒトp53エクソンに置き換えられた。 S47マウスの大部分は、WT p53マウス(48)とは異なり、年齢の12と18ヶ月の間に自発的に様々な組織型、特に肝臓癌の腫瘍を開発しました。, マウス胚性線維芽細胞およびヒトリンパ芽球細胞株では、S47変異体は、シスプラチンおよび他の遺伝毒性ストレスに応答して障害プログラム メカニズム的には、S47変異体は、Gls2(グルタミナーゼ2)およびSco2(48)などの代謝に関与する遺伝子のトランスアクティブ化のために欠陥があります。 フェロプトーシスにおけるGls2の役割と一致して、このグループは、S47細胞が著しくフェロプトーシス誘導剤エラスチンとRSL3(47、48)に耐性であったことが この欠陥は、s47マウスで観察された腫瘍を起こしやすい表現型に寄与する可能性があります。,
変異p53はFerroptosisに腫瘍細胞を感作
野生型p53は負のferroptosisに対する感受性を阻害するシスチン輸入SLC7A11の発現を調節する(5)。 この調節は正常細胞、腫瘍細胞において起こるが、SLC7A11の他のメディエーターは、この遺伝子の調節において優勢であるようである。 例えば、マスター antioxidant転写因子NRF2はまた、転写レベルでSLC7A11の発現を調節することができ、NRF2は、フェロプトーシスに対する癌細胞の保護におけるキープレーヤーとして関与している。, 例えば、肝細胞癌細胞におけるNRF2の阻害は、In vivoでのエラスチンおよびソラフェニブの抗癌活性を増加させる(56)。 P53の変異型は、直接相互作用によってNRF2機能を阻害することができ、あるグループは、変異p53を有する腫瘍がSLC7A11の非常に低いレベルを含み、したがって、フェロトーシスに対する感受性の増加を示すことがわかった。 特に、変異p53モデルにおけるSLC7A11の過剰発現は、SLC7A11発現のレベルは、変異p53駆動癌フェロプトーシス誘導化合物(57)を標的とするときに考慮されなければならないことを示唆し、薬剤耐性につながった。, この前提をサポートするために、p53の突然変異または欠失が頻繁なイベントである結腸直腸(CRC)癌における最近の研究は、変異体p53を保有するヒトCRC細胞株がWT p53とCRC細胞と比較してエラスチンを介した細胞死にはるかに敏感であることを示した。 これらの知見を検証するために、彼らは、P53ホットスポット変異のhct116とSW48細胞の両方におけるノックインがエラスチンに対する感受性を回復することを示した(51)。 これらのデータを見た新しいメカニズムによる癌転変異型p53で活用できる対象の治療すること。,
結論
代謝におけるp53の役割は非常に明確であり、おそらく直感的に明らかである:WT p53は、グルコース代謝および脂質合成を制限するが、変 P53による腫瘍抑制、および腫瘍の進行を駆動する変異体p53の能力に対するその代謝的役割の寄与は、明白に証明されているままである。 フェロトーシスの調節におけるp53の役割、および腫瘍抑制に対するこの機能の寄与は、さらに明らかではない。, マウスモデルからの説得力のあるデータは、p53がフェロプトーシスに対する細胞の感受性を調節するという前提をサポートしているが、これは自発的な腫瘍の発達を抑制するために基底p53の能力に制限される可能性があり、癌遺伝子ストレスマウスモデルでは、老化とアポトーシスが支配的な役割を果たすことは明らかである。 同様に、p53は、細胞型特異的方法でフェロプトーシス感受性を調節することができる。 異なったティッシュのferroptosisへの注意の動物モデルのより多くの調査は、腫瘍の抑制のferroptosisそしてferroptosisに於いてのp53の役割をもっと十分に理解するためにされる必要があります。, さらに、p53標的遺伝子がフェロプトーシスに対する感受性において役割を果たすものの明確なアイデアを達成する必要がある。 これらの質問の解決は、変異p53を有する腫瘍と戦うために多くの必要な新規の道を提供する必要があります。
著者の貢献
KG、SB、TB、AB-K、C-PK、およびMMはそれぞれこの記事の一から二の段落を書きました。 KGとSBは図をしました。 KGとMMの章を概説しました。,
利益相反声明
著者らは、この研究が潜在的な利益相反と解釈され得る商業的または財務的関係がない場合に行われたと宣言している。
レビュアー OAFとハンドリングエディターは、彼らの共有所属を宣言しました。
謝辞
この出版物で報告された研究は、受賞番号CA102184(MM)、CA201430(MM)、TL1TR002344(C-PK)、およびT32CA009171(TB)の下で国立衛生研究所によって支持された。, コンテンツは著者の責任のみであり、必ずしも国立衛生研究所の公式見解を表すものではありません。
3. ハンプトンTJ、ヴースデンKH。 P53による細胞代謝および低酸素症の調節。 コールド-スプリング-ハーバード(2016) 6(7):211-30. doi:10.1101/cshperspect.a026146
CrossRef Full Text|Google Scholar
10. 張C、劉J、呉R、梁Y、林M、劉J、ら。, 腫瘍サプレッサーp53負のターゲットRRADを介して低酸素症によって刺激解糖を調節します。 オンコターゲット(2014) 5(14):5535-46. doi:10.18632/oncotarget.2137
PubMed Abstract|CrossRef Full Text|Google Scholar
17. イ-ム、ユン-ジョン、ジョン-ジョン、 解糖とミトコンドリア酸化の間の代謝相互作用:逆ウォーバーグ効果とその治療的意味。 ワールドJバイオレットケム(2015) 6(3):148-61. 土井:10.4331/wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., 新規p53標的遺伝子カルニチンパルミトイルトランスフェラーゼ1Cの枯渇は、神経線維腫症i型腫瘍モデルにおける腫瘍増殖を遅らせる。 細胞死の違い(2013) 20(4):659-68. 土井:10.1038/cdd.2012.168
PubMed Abstract|CrossRef Full Text|Google Scholar
30. Ettinger SL,Sobel R,Whitmore TG,Akbari M,Bradley DR,Gleave ME,et al. 男性ホルモンの独立への進行の間の前立腺癌のステロールの応答の要素結合タンパク質そして下流のエフェクターのDysregulation。, Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL、Yang Q、Tong WM、Hergenhahn M、Wang ZQ、Hollstein M.キメラヒト/マウスp53遺伝子を有するノックインマウスは正常に発達し、DNA損傷剤に対する野生型p53応答を示す:新しい生物医学研究ツール。 がん遺伝子(2001) 20(3):320-8. 土井:10.1038/sj.オンク1204080
PubMed Abstract|CrossRef Full Text|Google Scholar
57. Liu DS,Duong CP,Haupt S,Montgomery KG,House CM,Azar WJ,et al. システムxC-/グルタチオン軸を阻害することは、選択的に変異p53蓄積を有する癌を標的とする。, Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















