Genome Spot (日本語)
経路解析は、特に遺伝子発現の研究において、バイオインフォマティクスにおいてこのような一般的な手順となっている。 最近の論文の調査を見ると、それを行うことができる方法がたくさんあるようです。 この記事では、一般的に使用される二つのツール、DAVIDとGSEAの違いについて説明します。

二つのアルゴリズムの背後にある概念は非常に異なっています。, DAVIDは、ユーザーが提供する遺伝子リストとキュレーションされたデータベース間の重複を判断し、ランダムな偶然によって予想されるものよりも大きな重複を 実験で考慮/検出されたすべての遺伝子を含むバックグラウンドファイルを提供することで、アルゴリズムの精度を向上できます。 ユーザが提供する遺伝子セットは、通常、有意性閾値を通過する遺伝子を選択することによって生成される。 DAVIDプロシージャは、創意工夫、AmiGO、GeneGOなどの他のものと似ています。, 有意性値の選択はほとんど任意であるが、FDR調整されたp<0.05で閾値を設定するのが一般的である。 しきい値を変更すると、濃縮結果が大幅に変更されます。 用いられる統計的検定には、超幾何学、フィッシャーの正確およびカイ二乗が含まれる。
任意の閾値を通過する遺伝子のリストを必要とする上記の方法とは対照的に、GSEAは統計アルゴリズムのすべてのデータポイントを使用するツールで “古典的な”方法では、遺伝子は、最もアップレギュレーションされたものから最もダウンレギュレーショ, ランクメトリック自体は異なりますが、符号付きp値を使用するか、折り畳み変更の90%信頼区間を下げることが有効な方法です。 の基礎試験を評価するか否かの遺伝子セットが豊かにする。 濃縮を試験するために、GSEAは、プロファイルの順列を行い、p値を経験的に推定するために千回以上設定された遺伝子の濃縮を計算する。 プロファイル全体を使用する他のツールは、Wilcoxon検定やKolmogorov-Smirnov検定などの分析試験を実行します。,
アルゴリズムの主な違いを理解したので、これら二つの手法の結果を比較し始めることができます。 私は前に議論したデータセット、RNA-seqによってプロファイルされたAML3細胞におけるアザシチジン治療を再訪しました。 DESEQによって生成された差分RNA-seq発現データを開始点として使用して、DAVIDとGSEAを比較します。
DAVIDのために、実験で検出されたすべての19,030個の遺伝子からなるバックグラウンドファイルを作成しました検出閾値を超える。 重要な(FDR<0.,05)up(2502遺伝子)およびdown(2907遺伝子)方向の遺伝子リストは、KEGG経路のみに焦点を当てた分析のためにDAVID6.7に別々に提出されました。
GSEAについては、先に説明したのと同じランクメトリック手順を使用しました。 私はgsea2-2.2.2で古典的なGSEAアルゴリズムを使用しました。msigdb v5.1のKEGG遺伝子セットを使用したjar実行可能ファイル。
下のグラフでは、統計学的に有意な遺伝子セット(FDR<0.05)の重複を上下調節経路について別々に決定しました。, 私はまた、背景が提供されていないときにDAVIDの結果の交差点を含めました(好奇心のためだけで、推奨される手順ではありません)。
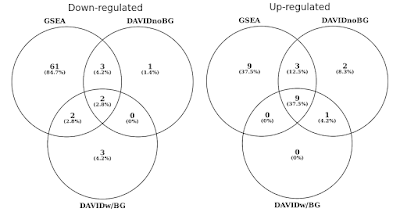
同じ遺伝子発現プロファイルを使用してデイビッド対GSEA。 数値は、有意に濃縮された遺伝子セットを表す(FDR<0.05)。
あなたは、上下の規制方向の両方で、GSEAはDAVIDと比較してより重要な結果を識別することに気づくでしょう。 私はこれがREACTOMEやGOなどの他の遺伝子セットライブラリの場合でもあることを発見しました。, 二つのツール間の感度の違いは、DAVIDがすべてのデータポイントを使用するGSEAに対して、遺伝子の離散リストを使用して、異なるアルゴリズムに帰着します。 GSEAは、発現の微妙な変化を受けている多くのメンバーを有する遺伝子セットを同定する際にはるかに敏感になるであろう。 個別に、これらの変化は統計的に有意ではないが、集合内の遺伝子の大部分が集合的に発現にシフトすると、その傾向は統計的に非常に有意であり得る。,
より敏感なアルゴリズムに加えて、DAVIDの遺伝子セットと遺伝子注釈は本当に古くなっています。 DAVIDの多くのデータソースの最後の更新は、2009年半ばから後半にあります。 したがって、Ensemblからの多くの遺伝子アクセッションは支持されない可能性がある。 確かに、19,030を含む私の背景ファイルから、13,488だけがDAVIDによって認識されました。 DAVIDがまだ受け取っている引用の数が増え続けているように、更新された遺伝子セットの欠如も懸念されています。,
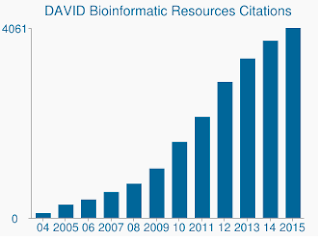
DAVIDは、基礎となるデータが悲惨に古くなっているにもかかわらず、年間何千もの引用を受け取っています。結論として、研究者は、重複分析のために遺伝子セットを提出するのではなく、濃縮(GSEA、CAMERA、WilcoxGST、Pathifier)を決定するためにプロファイル全体を見るツールを使用する DAVIDを使用すると、迅速かつ簡単なオプションかもしれませんが、これは感度の大幅な低下を犠牲にして来ます。
さらに読む
JAティモンズ、KJ Szkop、IJギャラガー。, バイアスの複数のソースは、グローバルオミクスデータの機能濃縮分析を混乱させる。 ゲノム生物学、2015年。 16:186
リナ-ワディ、モナ-マイヤー、ジョエル-ワイザー、リンカーン-D-スタイン、ジュリ-ライマンド パスウェイ濃縮分析に対する知識蓄積の影響。 バイオルクシウ doi:http://dx.doi.org/10.1101/049288
編集:月に2016,DAVIDは更新されました!
https://david.ncifcrf.gov/content.jsp?file=release.html















