국경 내분비학
소개
종양 억제 유전자 TP53 되었습니다 가장 크게 공부한 인간의 유전자의 발견 이후 약 40 년 전(1). 이 상태 뒤에있는 주된 이유는 p53 이 암 발병을 예방하는 데 중요한 역할을하며”게놈의 수호자로 널리 간주됩니다.,”약간의 시간에 대한 일반적으로 믿고 그 p53 의 역할에 종양 억제에 대해 그것의 기능을 유도하멸,세포주기 체포하고,노화의 암 세포는(2)입니다. 그러나,그것은 이제 점점 더 명확 p53 을 조절하는 많은 다른 경로 셀에서는 이러한 다른 경로에도 역할을 재생할 수 p53 의 능력을 기능으로 종양 억제기(3). 특히,신진 대사와 ferroptosis 에 관여하는 유전자의 조절에서 p53 의 역할은 종양 발달을 억제하는 능력에 연루되어있다., Ferroptosis 새로운 세포의 죽음을 통로 첫 번째 특징으로 2012 년 최고의 수명은 철-의존,caspase-독립적인 형태로 세포 죽음의 구동에 의해 형성의 과산화 지방(4). 특히,두 마우스 모델에 포함된 설계에 돌연변이 p53 을 제거하는 능력의 p53apoptosis 유도하고 노화가 모두 유지하는 능력을 억제하는 자발적인 종양 개발에 이러한 돌연변이 유지하는 능력을 transactivate 유전자에서의 대사과 ferroptosis(5,6)., 신진 대사 및 ferroptosis 의 조절에 p53 을 암시하는 데이터의 요약은 아래에 자세히 설명되어 있습니다.
야생형(WT)p53 조절 긍정적으로 산화 인 산화 억제 포도당 대사
Wild-type p53 조절 변화의 다양성에 의해 세포를 선호하는 미토콘드리아의 호흡을 통해 해당 분해를 통해,부분 transactivation SCO2(산화효소 시토크롬 c 어셈블리),재생하는 직접적인 역할을에서 산화 인 산화(7)입니다., p53 은 또한 GLS2(Glutaminase2)의 transactivation 을 직접 조절한다;이 효소는 미토콘드리아(8)의 에너지 원으로서 글루타민 사용을 허용한다. 또한,WT p53 은 포도당 전달체 GLUT1 및 GLUT4 를 전사적으로 억압하고 rrad 및 TIGAR 를 transactivating 함으로써 당분 해제를 부정적으로 조절한다;둘 다 당분 해의 억제제이다(9-11). 마지막으로,p53 은 또한 효소 포도당-6-인산 탈수소 효소에 직접 결합하고 억제하여 포도당 대사를 억제합니다(12)., 정상,스트레스를받지 않는 유기체에서 p53 이 세포에서 대사 상태를 직접 조절한다는 것은 이들 및 다른 연구에서 분명합니다(그림 1). 놀랍지 않게도,이 유전자와 그 조절 자의 대부분은 비만과 당뇨병을 포함한 대사성 질환에 연루되어있다(13).
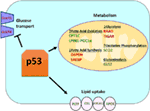
그림 1. 신진 대사에서 야생형(WT)p53 의 역할. P53 에 의해 긍정적으로 조절되는 유전자는 녹색으로 표시되고 p53 에 의해 부정적으로 조절되는 유전자는 빨간색으로 표시됩니다., p53 은 지질 흡수,지방산 산화,산화 적 인산화 및 글루타민 분해를 촉진하면서 포도당 수송,당분 해 및 지방산 합성을 억제합니다.
돌연변이 p53 조절 긍정적으로 바르 부르크 대사(호기성 분해)
에 대비하는 함수의 WT p53,돌연변이 p53 에서 종양 세포 호의 호기성 분해에 의해 부분적으로,강화 인신매매의 포도당이 수송 GLUT1 플라즈마 멤브레인,따라서 증가하고 포도당 가져오기(14,15)., P53 의 돌연변이에 따라,SCO2 와 GLS2 의 감소 된 수준과 GLUT1 과 GLUT4 의 증가 된 수준은 산화 적 인산화에 비해 호기성 당분 해를지지한다. 이러한 방식으로,돌연변이 p53 에 기여 하는 것으로 성향의 종양세포 활용하는 호기성 분해에서의 호의 산화 인 산화나라 바르 부르크 대사(15). 의 특징 중 하나는 암 규제 완화의 대사,일반적으로 증명에 의해 이치에서 호기성 분해하는 산화 인 산화., 하지만 이것이 낮고 덜 효율적인 ATP 수익률,그것은 믿고 암세포 이익으로 전환 당분 중간체 생합성 경로를 위해 필요한 신속한 세포분열(16). 이사 스위치 또한 이끌어 줄 미토콘드리아-mediated apoptosis 고 더 효율적 신호를 사용할 수 있는 대사 산물에서 암세포(17).
일반적인 유전자 변형 TP53 영향에서의 기능에 대
일반적인 코딩은 지역의 다형성 p53codon72,인코딩 중 하나에 대한 프롤린(P72)또는 아르기닌(R72)., 이 아미노산 변이는 스트레스 후 세포 운명과 관련하여 p53 기능에 영향을 줄 수 있습니다. DNA 손상에 대한 반응으로,p53 의 P72 변형은 주로 세포주기 정지를 유발하는 반면,R72 변형은 주로 세포 사멸 또는 세포 사멸을 유도한다(18,19). 이러한 기능의 차이에도 불구하고,코돈 72 변이는 암 감수성과 일관되게 연관되어 있지 않았다(20). 대조적으로,인간 연구에서이 다형성은 체질량 지수와 당뇨병의 위험 증가와 유의 한 관련이 있습니다(21,22)., 이제는 지원되는 연구에 의해서 쥐는 곳에,마우스 모델에 대한 이 codon72 개 쇼 증가 높은 지방 다이어트 유도 당뇨병에서 마우스로 R72 변종,비교하 P72. 이 연구에서 p53 표적 유전자 TNFa 와 NPC1L1 은 R72 마우스에서식이 유발 비만의 증가에 중요한 조절 자로 확인되었다(23). 흥미롭게도,R72 변이체는 또한 영양소 박탈에 반응하여 세포의 증가 된 생존을 부여하는 것으로 나타났다(24)., 이러한 연구 결과가 주도하는 가설 R72 변종의 p53 일어나 선정되었으로 인구를 마이그레이션,북쪽에는 추운 날씨가 증가를 필요로 하는 지방이 축적이지만 생존에 응답하는 영양소의 부족 또한 것에서 선택(24).
p53 조절 지질 대사
지만 p53 은 잘 알려진 조절하는 분해 및 구연산 사이클,p53 도를 보여왔는 역할을 규제하는 지질 대사(25)., WT p53 은 지방산 합성을 억제하면서 지방산 산화를 향상시켜 지질 합성의 음성 조절제 역할을한다고 믿어진다(25). 지질 대사에 역할을하는 몇 가지 p53 표적 유전자가 있습니다. Sanchez-Macedo 와 동료들은 카르니틴 팔미 토일 트랜스퍼 라제 1C(CPT1C)가 p53 에 의해 전사 적으로 조절된다는 것을 입증했다;이 효소는 활성화 된 지방산을 미토콘드리아로 운반하는 것을 돕는다., 에서 지원의 역할에 대한 이 p53-제 유전자,암이 그룹 보이는 Cpt1c 불충분 한 쥐 디스플레이 지연되는 종양의 개발 및 높은 생존율(26). Lipin1(LPIN1)은 또 다른 p53 표적 유전자이다;LPIN1 은 적절한 지방 세포 발달에 필요하며 낮은 영양 조건 하에서 유도된다(27). Finck 및 동료에게 보여주었 LPIN1 와 상호 작용 PGC-1α,다른 알려진 p53 대상 유전자의 역할에 대사,그리고 그 이 상호 작용이 활성화하는 유전자의 표현에 관련된 홍보 지방산 산화(28).,
외에 바로 조절하는 유전자의 전사에 관련된 지질 대사,p53 할 수 있도 조절 지질 대사는 방식으로 포함하는 직접적인 단백질–단백질 상호 작용입니다. 예를 들어,glucose-6-phosphate dehydrogenase,이는 속도 제한하는 효소에서 pentose 인산 경로,결합 하고 직접에 의해 저해 p53,결과적으로 NADPH 생산은 결과적으로 감소 지방산 합성(12)., 린 규제 요소인 단백질(SREBP)가족의 전사 요인 조절현에 관련된 유전자의 콜레스테롤,지방산,triacylglycerol,인지질과 합성(29-31). WT p53 은 srebp 기능(32)을 억제하는 반면,p53 의 돌연변이 형태는 SREBP 에 직접 결합하여 전사 기능을 향상시켜 인간 종양에서 srebp 활성을 증가시킵니다(33,34). 결과적으로,돌연변이 p53 은 인간 유방 종양에서 스테롤 생합성 유전자의 높은 발현과 상관 관계가있다(34,35)., 마지막으로,AMP-activated protein kinase(AMPK)효소 활성화 되는 아래에서 낮은 영양소 레벨 또는 에너지는 긴장하고 알려져 있을 억제하는 지방산 합성과의 상호 작용에 의해 acetyl-CoA-carboxylase 및 SREBP-1(36,37). Zhou 와 동료들은 돌연변이 p53 이 AMPK 에 우선적으로 결합하고 억제하여 증가 된 지방산 합성을 유도한다는 것을 보여 주었다. 결과적으로,돌연변이 p53 단백질은 증가 된 AMPK 신호 전달을 유도하여 종양 세포의 침습성 세포 성장에 기여한다(33). 덜 탐구 된 영역은 지질 수송에서 p53 의 역할입니다., P53 이 아포지 단백질 B(apoB)와 apoB 편집 효소 복합체 1 을 전사적으로 조절하여 아테롬성 지단백질 조절에서 p53 의 역할을 나타내는 것으로 나타났다(38). 마이크로어레이 분석의 인간의 간 유래 세포 식별 인지질전달,단백질 ATP binding cassette A12 및 에스테르 카르복실기 lipase 으로 세 p53 대상 유전자는 모든 역할을에서 지질의 수송(39,40)., 전반적인,하지만 그것은 분명 p53 중요한 역할을 중재에 지질 합성 및 신진대사,의 기여를 이 경로,그리고 이러한 p53 대상 유전자,종양 억제에 의해 p53 확인할 수 있습니다(그림 1).
Ferroptosis 새로운 세포의 죽음을 통로 구동에 의해 지질 과산화
2012 년에 딕슨과 동료 발견한 소설의 양식을 규제한 세포는 죽음이라는 ferroptosis. Ferroptosis 는 산화 된 지질의 축적으로 인한 철 의존성,카스파 제 독립적 인 세포 사멸 형태이다(4,41)., 이 프로세스에 의해 구동되는 비활성화의 글루타티온 peroxidase4(GPX4),효소를 담당하는 변환 치명적인 지질 hydroperoxides 비독성 지질 알코올,필요로하는 글루타티온기능(41). 고도 불포화 지방산(pufas)의 과산화가 ferroptosis 에 의한 세포 사멸의 원동력이라고 믿어진다. PUFAs 포함 bis-allylic 양성할 수 있는 추출 및 생산 활성산소는 것이 산소와 반응을 만들고,더 활성산소와 연쇄 반응 결과의 지질 reactive oxygen species(42)., 정확한 메커니즘의 휴 죽음으로 ferroptosis 알 수 없는 남아 있지만,하나의 가설은 지질 손상 파괴에 이르게 플라즈마의 막(43). 고 있는 것으로 추측 ferroptosis 수 있는 메커니즘의 종양 억제에 의해 작동하는 세포를 제거하는 영양 박탈하였거나 노출된 환경적 스트레스 또한 감염.,
약리학적인 규제의 Ferroptosis
Ferroptosis 수 있습 유도를 사용 억제제 시스템 xc−과 같은 erastin,또는 아날로그와 같은 글루타민산염과 sorafenib,을 억제하의 수입 cystine 결과,고갈 글루타티온과 이후의 비활성화 GPX4. 또한,ferroptosis 에 의해 유발 될 수 있습니다(1S,3R)-RSL3(이하 RSL3)에 직접 결합을 억제 GPX4(4,5,42). Buthione sulfoximine,FIN56,FINO2,CCl4 및 cisplatin 은 세포에서 ferroptosis 를 유도하는 것으로 입증 된 다른 약제입니다., 에 의해 죽음을 ferroptosis 을 방지할 수 있을 억제하여 지질 과산화를 사용하여 수행할 수 있습지는 항산화제와 같은 ferrostatin-1,liproxstatin-1,또는 비타민 E 철 킬레이트와 같은 deferoxamine 또는 cicloprox 은 또 다른 도구를 억제하는 데 사용됩 ferroptosis 의 수준을 감소시켜서 철이 있습니다. PUFAs 를 고갈 시키거나 단일 불포화 지방산을 세포 배양 배지에 첨가하면 ferroptosis 에서 세포를 구할 수 있습니다(42,44).,
Ferroptosis 에 연루 p53-Mediated 종양 억제
2012 년에 구하고 동료들이 개발된 마우스 모델에는 세 가지 일반적으로 acetylated lysine 잔류물에서 DNA-binding domain of p53 돌연변이 되었 아르기닌,따라서 할 수 없습 acetylated;이것은 마우스로 불리 3KR 마우스입니다. 특히,세포에서 3KR 마우스 있을 받을 수 없습 p53-dependent apoptosis,세포주기 체포하거나,노화,그리고 실제로 3KR 돌연변이 p53 실패 transactivate 대부분의 p53 대상 유전자입니다., 흥미롭게도이 마우스 모델은 자발적으로 암을 발생시키지 않아 p53 이 노화 또는 세포 사멸과 독립적 인 종양 발달을 억제 할 수 있음을 암시합니다(45). 이 그룹이 있는 돌연변이 3KR 단백질을 유지하는 능력을 받 ferroptosis 고 통제 시스틴은 대사 조절의 식스틴 수입 SLC7A11;이 제안하는 ferroptosis 중 하나가 될 수 있는 통로의 기초가 되는 p53-mediated 종양 억제., 면 야생의 유형 및 3KR MEFs 었으로 처리 ferroptosis 유도 Erastin,거의 50%의 세포가 죽었을 관찰하는 반면 p53null MEFs 전시 20%세포 사망이니 이는 p53sensitizes 세포 ferroptosis,또는 다른 키 레귤레이터 역할을 ferroptosis(5). 그 후,구와 동료를 식별한 추가 acetylation 사이트에서 리신의 98p53,그리고 그들은 생성된 마우스 모델에는 모든 네 acetylation 사이트는 돌연변이 아르기닌(4KR)., 흥미롭게도,4KR 돌연변이었을 조절할 수 없습에 관여하는 유전자 ferroptosis 다음과 같 SLC7A11,그리고와 달리 3KR 돌연변이었을 억제할 수 없는 종양 개발(46). 현재 상관 관계가 있지만,이러한 데이터는 종양 발달을 억제하는 능력에서 ferroptosis 에서 p53 의 역할을 암시합니다.
에서 비 변형 셀,p53 긍정적으로 조절 Ferroptosis
이외에 SLC7A11,여러 다른 직접 p53 대상 유전자가 발견되었하는 역할을 ferroptosis. 여기에는 GLS2,PTGS2 및 SAT1 이 포함됩니다., 두 개의 개별 그룹의 연구는 글루타티온을 감소시키고 세포 ros 수준을 증가시키는 것으로 알려진 ferroptosis 에서 GLS2 의 역할을지지합니다. 강 및 동료에 사용 ferroptosis 억제물과 결합된 glutaminolysis 억제물을 억제하는 Erastin-유도 ferroptosis 함으로써 보여주는 ferroptosis 필요 glutaminolysis 및 GLS2(47). 머피와 동료들을 보였는 다형성의 변 p53 할 수 있었을 유도하는 성장을 체포하고 노화에서는 인간과 murine 세포이지만 실패했을 억제 SLC7A11 또는 transactivate GLS2., 이 변이체는 ferroptosis 를 유도하고 종양 발달을 억제하는데 현저하게 손상되어 ferroptosis 매개 종양 억제에서 p53 의 역할을 다시 암시했다(48). Ferroptosis 에서 역할을하는 또 다른 p53 표적 유전자는 효소 cyclooxygenase-2 를 암호화하는 유전자 인 PTGS2 입니다. Stockwell 과 동료들은 Erastin 과 RSL3 을 이용한 ferroptosis 의 유도가 PTGS2 의 상향 조절을 유도한다는 것을 처음으로 보여 주었다(41). 주목할 만하게,PTGS2 는 p53-null 세포에서 ferroptosis 유도제에 의해 상향 조절되지 않았으며,이 조절이 p53 의존적임을 시사한다(5)., 현재,PTGS2 의 상향 조절은 ferroptosis 마커(5,41)로 널리 사용됩니다.
gu 그룹의 최근 연구에 따르면 p53 표적 유전자 SAT1 은 ferroptosis 를 조절합니다(49). 저자 식별 sat1 세계로 직접적인 대상의 p53 보는 침묵의 sat1 세계 줄일 세포의 죽음에 의해 유도된 반응성산소는 세포에서 WT p53 지만,아무 효과가 없었에 p53-null 세포입니다. Mechanistically,이는 그룹 보이는 sat1 세계 증가시킨 수준과 활동의 arachidonate15-lipoxygenase,철-바인딩하는 효소를 산화 PUFAs 및 과산화 지방이 증가., 주목할 만하게,이 연구는 p53 이나 SAT1 단독으로 ferroptosis 를 유도하기에 충분하지 않은 것으로 보이지 않는다는 것을 보여 주었다. 대신,결합된 데이터는 일관성이 있음을 전제로는 p53,의 미덕에 의해 규제에 기여 하는 유전자 ferroptosis,조절감도의 세포이 통로,직접 보다는 오히려 유도하는 ferroptosis. P53 이 ferroptosis 에 관여하는 다른 유전자를 조절하는지 여부는 여전히 결정되어야한다(그림 2).
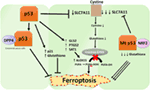
그림 2. Ferroptosis 에서 p53 의 다양한 역할., 억제의 글루타티온 peroxidase4(GPX4),키 효소는 촉매 변환기의 불포화 지방산(PUFAs)을 포함하는 과산화물,알코올,은 키 드라이버 ferroptosis. 문맥에 따라 p53 은 ferroptosis(대장 암 세포에서와 같이)를 억제하거나 ferroptosis 를 촉진 할 수 있습니다. 돌연변이 p53 은 야생형 p53 보다 훨씬 더 ferroptosis 에 세포를 민감하게합니다.,
부세포,p53 부정적인 조절 Ferroptosis
연구는 최근에 출판 Tarangelo 와 동료는 것을 보여줍 p53 부정적인 조절 ferroptosis 에서 암세포(50). 이 그룹은 p53 을 안정화시키는 화합물 인 Nutlin-3 으로 세포를 전처리하는 것이 여러 세포 유형에서 ferroptosis 의 발병을 지연 시킨다는 것을 발견했다. Ferroptosis 의 지연된 발병은 중요한 p53 전사 표적 인 CDKN1A(인코딩 p21)에 의존하는 것으로 밝혀졌다., 메커니즘을 통해 어떤 p21 지연 ferroptosis 은 아직 밝혀지만,그것은 믿고 보호의 세포내는 글루타티온에 기여 요인이 될 수 있습니다 감소를 위한 ferroptosis 니다. 저자는 결론을 내릴 p53–p21 축할 수 있습 암세포 생존하는 조건에서의 대사 등의 스트레스 시스틴 박탈에 의해,억제의 발병 ferroptosis(50). 최근의 한 연구는 p53 을 억제 ferroptosis 에서 대장암 세포에 바인딩하여 효소의 디펩-펩티-4(DPP4),는 변조기의 ferroptosis 및 지질 대사가 있습니다., 기계적으로,이 연구는 p53 이 핵 효소 비활성 풀에서 dpp4 를 격리시킴으로써 ferroptosis 를 길항한다는 것을 보여 주었다. 의 부재에서 p53,DPP4 무료과 상호 작용하는 복합물을 형성과 NOX1;이 증가로 이어집 지질 과산화 및 ferroptosis. DPP4 의 억제는 ferroptosis 를 유의하게 억제하는 반면,DPP4 의 과발현은 특히 p53-고갈 된 세포에서 에라 스틴 감도를 유발한다(51). 전사 의존적 및 전사 독립적 메커니즘을 통한 p53 에 의한 ferroptosis 의 양방향 제어는 문맥 또는 세포 유형 의존적 일 수있다(그림 2).,
이 P47S 다형성의 TP53 영향을 미치 Ferroptosis 및 종양 억제
이외에 missense 돌연변이,거기에 몇 가지 기능적으로 중요한 단 뉴클레오티드 polymorphisms(SNPs)에 TP53 유전자 및 기타 단백질 규제 알려진이 경로(같은 MDM2 및 MDM4). Pro47Ser 변이체(이하 S47)는 단백질의 아미노산 서열을 변화시키는 p53 코딩 영역(Pro72Arg 이후)에서 발견되는 두 번째로 흔한 SNP 이다., 더 나은 명료의 영향을 이 변종에 p53 기능 및 암 위험,머피는 그룹을 생성을 인간답게 p53 노크-마우스 모델에는 exons4-9murine p53 에 의해 대체되었다 인간 p53exons 하나를 포함하는 야생의 유형 또는 S47 변형(52-55). 대부분의 S47 쥐 자발적으로 개발한 종양의 다양한 조직학,특히 간암,간 12 18 개월과는 달리,WT p53 마우스(48)., 마우스 배아 섬유아세포와 위대한 약속을 보유 하고있는 인간의 세포 라인 S47 변형을 보여 장애로 반응하 cisplatin 과 다른 차적인 스트레스입니다. 기계적으로,S47 변이체는 Gls2(glutaminase2)및 Sco2(48)와 같은 대사에 관여하는 유전자의 transactivation 에 결함이 있습니다. Ferroptosis 에서 Gls2 의 역할과 일치하여,이 그룹은 s47 세포가 ferroptosis 유도제 인 Erastin 과 RSL3 에 현저하게 내성이 있음을 발견했다(47,48). 이 결함은 s47 마우스에서 관찰 된 종양이 발생하기 쉬운 표현형에 기여할 수있다.,
돌연변이 p53 은 종양 세포를 Ferroptosis
야생형 p53 에 민감하게하여 ferroptosis(5)에 대한 민감성을 억제하는 시스틴 수입자 SLC7A11 의 발현을 부정적으로 조절한다. 이 조절은 정상 세포에서 발생하지만,종양 세포에서는 SLC7A11 의 다른 매개체가이 유전자의 조절에서 우세한 것으로 보인다. 예를 들어,항산화제 마스터 전사 인 NRF2 할 수 있도 조절 식 SLC7A11transcriptional 수준에서,그리고 NRF2 내포되었습니다에서 핵심적 보호를 암세포에 대한 ferroptosis., 예를 들어,간세포 암 세포에서 NRF2 의 억제는 생체 내 에라 스틴과 소라 페닙의 항암 활성을 증가시킨다(56). 돌연변이 형태의 p53 을 억제할 수 있 NRF2 기능에 의해 직접적인 상호작용,그리고 한 그룹이 있는 종양으로 돌연변이 p53 포함한 매우 낮은 수준의 SLC7A11,따라서 보여 감도 증가하 ferroptosis. 특히,overexpression SLC7A11 에서 돌연변이 p53 모델 led 약물 저항,제안하는 수준의 SLC7A11 식이 고려되어야를 대상으로 하는 경우 돌연변이 p53 기반 암으로 ferroptosis-유도하기화합물(57)., 의 지원에 이내,최근 작업장(CRC)암,돌연변이 또는 삭제 p53 은 자주 이벤트,보여주는 인간의 CRC 세포 라인을 품고 돌연변이 p53 었다 훨씬 민감하 Erastin 중재 세포가 죽을 때에 비해 CRC 세포 WT p53. 을 확인 이러한 연구 결과는,그들이 보여주는 노크에서의 p53 핫스팟을 돌연변이에서 모두 HCT116 및 SW48 세포로 복원에 감도 Erastin(51). 이 데이터는 돌연변이 p53 에 의해 구동되는 암이 표적 치료법을 사용하여 악용 될 수있는 새로운 메커니즘을 강조합니다.,
결론
의 역할을 p53 에서 대사가 매우 명확하고 어쩌면 직관적으로 분명:WT p53 한 포도당 대사와질 합성하는 동안,돌연변이 p53 나타납하여 반대를 할 수 있습니다. 의 기여 대사 역할을 종양 억제에 의해 p53,고의 능력을 돌연변이 p53 드라이브 종양 진행 남아 있는지 명백히 입증합니다. Ferroptosis 의 조절에서 p53 의 역할 및 종양 억제에 대한이 기능의 기여는 훨씬 덜 명확합니다., 는 매력적인 데이터 마우스 모델을 지원하는 전제 p53 을 조절한 세포의 민감도 ferroptosis,이 제한될 수 있습 능력의 기초 p53 을 억제하는 자발적인 종양 개발에 oncogene-마우스조 모델,그것은 노화 및 apoptosis 플레이 주된 역할을합니다. 유사하게,p53 은 세포 유형 특이 적 방식으로 ferroptosis 감도를 조절할 수있다. 더 많은 연구에서 동물 모델에 대한 관심과 함께,ferroptosis 다른 조직에서 수행 할 필요가 더욱 완벽하게 이해하는 역할에서 p53ferroptosis 및 ferroptosis 에서 종양 억제., 또한,p53-표적 유전자가 ferroptosis 에 대한 민감성에 어떤 역할을하는지에 대한 명확한 아이디어가 달성 될 필요가있다. 이러한 질문의 해결은 돌연변이 p53 과 종양을 퇴치하기 위해 많이 필요한 새로운 길을 제공해야합니다.
저자 기고
KG,SB,TB,AB-K,C-PK 및 MM 각각은이 기사의 1~2 단락을 썼다. KG 과 SB 는 그림을했다. KG 과 MM 이 장을 설명했습니다.,
이해의 충돌 문
저자가 선언하는 연구가 수행되었의 부재에서 어떠한 상업 또는 금융 서비스를 제공하는 것으로 해석될 수 있는 잠재적인 이해의 충돌.
리뷰어 OAF 와 핸들링 편집자는 공유 제휴를 선언했습니다.
감사
연구보고 이 책에서 지원되었에 의해 국가기관의 건강 상호 CA102184(MM),CA201430(MM),TL1TR002344(C-PK),그리고 T32CA009171(TB)., 내용은 전적으로 저자의 책임이며 반드시 국립 보건원의 공식 견해를 나타내는 것은 아닙니다.나는 이것이 내가 할 수있는 유일한 방법이라고 생각한다. 험프 턴 TJ,Vousden KH. P53 에 의한 세포 대사 및 저산소증의 조절. 콜드 스프링 하브 Perspect 메드(2016) 6(7):211-30. doi:10.1101/cshperspect.a026146
CrossRef 전체 텍스트|Google Scholar
10. 장 C,리우 J,우 R,리앙 Y,린 M,리우 J,외., 종양 억제 p53 은 표적 RRAD 를 통해 저산소증에 의해 자극 된 당분 해를보다 부정적으로 조절합니다. 온코타겟(2014) 5(14):5535-46. doi:10.18632/oncotarget.2137
PubMed Abstract|CrossRef 전체 텍스트|Google Scholar
17. 이엠,윤제이. 당분 해와 미토콘드리아 산화 사이의 대사 상호 작용:역 와르 부르크 효과와 그 치료 적 의미. 월드 제이바이올 켐(2015) 6(3):148-61. 도이:10.4331/wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., 새로운 p53-표적 유전자 카르니틴 팔미 토일 트랜스퍼 라제 1C 의 고갈은 신경 섬유종증 i 형 종양 모델에서 종양 성장을 지연시킨다. 세포 사멸은 다릅니다(2013) 20(4):659-68. doi:10.1038/cdd.2012.168
PubMed Abstract|CrossRef 전체 텍스트|Google Scholar
30. Ettinger SL,Sobel R,Whitmore TG,Akbari M,Bradley DR,Gleave ME,et al. 안드로겐 독립성으로 진행하는 동안 전립선 암에서 스테롤 반응 요소 결합 단백질 및 다운 스트림 이펙터의 조절 장애., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., 루오 JL,양 Q,통 WM,Hergenhahn M,왕 ZQ,Hollstein M. 노크에 있는 쥐 chimeric human/murine p53 유전자의 정상적으로 개발 및 보 wild-type p53 응답 DNA 손상 에이전트:새로운 생명 연구 도구입니다. 온코겐(2001) 20(3):320-8. 도이:10.1038/sj.온씨.1204080
PubMed Abstract|CrossRef 전체 텍스트|Google Scholar
57. Liu DS,Duong CP,Haupt S,Montgomery KG,House CM,Azar WJ,et al. 시스템 xc-/글루타티온 축을 억제하는 것은 돌연변이-p53 축적으로 암을 선택적으로 표적화한다., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















