게놈소
경로 분석한 일반적인 절차에서의 생물정보학,특히에서 공부하고 유전자 발현. 당신이 최근 논문의 설문 조사를 보면 그것이 할 수있는 방법의 무리가있는 것 같습니다. 이 게시물에서는 일반적으로 사용되는 두 가지 도구 인 DAVID 와 GSEA 의 차이점에 대해 설명합니다.

개념 두 개의 알고리즘은 매우 다릅니다., 다윗을 결정 사이의 중복 사용자가 제공하는 유전자 목록 및 기획하는 데이터베이스,보고에 대한 중복되는 더 큰에 의해 예상되는 임의의 기회가있다. 실험에서 고려/검출 된 모든 유전자를 포함하는 배경 파일을 제공하여 알고리즘의 정확성을 향상시킬 수 있습니다. 사용자가 제공 한 유전자 세트는 일반적으로 유의 임계 값을 통과하는 유전자를 선택하여 생성됩니다. 데이비드 절차는 독창성,아미고 및 GeneGO 와 같은 다른 사람들과 유사합니다., 선택의 중요성 값은 크게 임의의,하지만 그것은 일반적인 임계 값을 설정하에서 루즈벨트 조정 p<0.05. 임계 값을 수정하면 농축 결과가 상당히 변경됩니다. 고용 된 통계 테스트에는 하이퍼 지오 메트릭,피셔의 정확한 및 카이 제곱이 포함됩니다.
에 대비하기 위한 방법을 요구하는 목록의 유전자 전달하는 임의의 임계값,GSEA 은 도구를 사용하는 모든 요소에서 통계 알고리즘이 있습니다. “고전적”방법에서 유전자는 가장 상향 조절 된 것에서 가장 하향 조절 된 것까지 순위가 매겨진다., 순위를 측정 자체가 다양하지만 두 가지 유효한 방법을 사용하여 서명 p-value,또는 낮은 90%신뢰 구간의 접 변경합니다. 시험의 기본은 유전자 세트의 구성원이 프로파일의 한쪽 끝에서 농축 된 것처럼 보이는지 여부를 평가하는 것입니다. 농축을 테스트하기 위해 GSEA 는 프로파일의 순열을 수행하여 p-값을 경험적으로 추정하기 위해 유전자 세트의 농축을 천 번 이상 계산합니다. 전체 프로파일을 사용하는 다른 도구는 Wilcoxon 및 Kolmogorov-Smirnov 테스트와 같은 분석 테스트를 수행합니다.,
이제 알고리즘의 주요 차이점을 이해 했으므로이 두 기술의 결과를 비교하기 시작할 수 있습니다. 나는 rna-seq 에 의해 프로파일 링 된 AML3 세포에서 azacitidine 처리 인 이전에 논의한 데이터 세트를 재검토했다. DESeq 에 의해 생성 된 미분 RNA-seq 식 데이터를 시작점으로 사용하여 DAVID 와 GSEA 를 비교할 것입니다.
DAVID 의 경우,검출 임계 값 이상의 실험에서 검출 된 모든 19,030 개의 유전자로 구성된 백그라운드 파일을 만들었습니다. 중요한(FDR<0.,05)up(2502 유전자)및 down(2907 유전자)방향의 유전자 목록은 분석을 위해 DAVID6.7 에 별도로 제출되었으며,KEGG 경로에만 초점을 맞추었다.
GSEA 의 경우 이전에 설명한 것과 동일한 순위 메트릭 절차를 사용했습니다. Gsea2-2.2.2 와 함께 고전적인 GSEA 알고리즘을 사용했습니다.MSigDB v5.1 의 KEGG 유전자 세트를 사용하는 jar 실행 파일.
아래 그래프에서,내 결정이 겹치의 통계적으로 중요한 유전자의 세트(FDR<0.05)별로 아래로 규제되는 경로., 또한 배경이 제공되지 않을 때 데이비드 결과의 교차점을 포함 시켰습니다(호기심에만 권장되는 절차는 아닙니다).
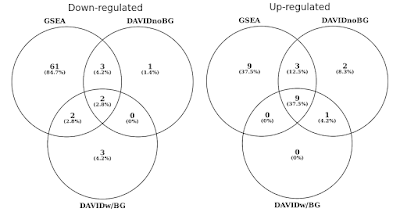
동일한 유전자 발현 프로파일을 사용하는 DAVID vs GSEA. 숫자는 상당히 농축 된 유전자 세트를 나타냅니다(FDR<0.05).
상향 및 하향 조절 방향 모두에서 GSEA 가 DAVID 와 비교하여 더 중요한 결과를 식별한다는 것을 알 수 있습니다. 나는 이것이 REACTOME 과 GO 와 같은 다른 유전자 세트 라이브러리에서도 마찬가지라는 것을 발견했다., 차이에서 감도를 사이에 두 개의 도구로 귀결을 서로 다른 알고리즘,데이비드를 사용하여 개별 유전자 목록,대 GSEA 사용하는 모든 데이터 지점입니다. GSEA 는 발현의 미묘한 변화를 겪고있는 많은 구성원을 가지고있는 유전자 세트를 확인하는 데 훨씬 더 민감 할 것입니다. 개별적으로,이러한 변경되지 않은 통계적으로 중요하지만,때 대부분의 유전자에서 설정한 집단적으로 변화를 표현하는 추세이 될 수 있는 아주 중요한 통계적으로 말합니다.,
더 민감한 알고리즘 외에도 DAVID 의 유전자 세트와 유전자 주석은 실제로 오래되었습니다. 데이비드의 많은 데이터 소스의 마지막 업데이트는 2009 년 중반에서 후반에 있습니다. 따라서 Ensembl 의 많은 유전자 가입은 지원되지 않을 수 있습니다. 실제로 19,030 이 포함 된 내 배경 파일에서 13,488 만이 DAVID 에 의해 인식되었습니다. 업데이트 된 유전자 세트의 부족은 또한 데이비드가 여전히받는 인용 횟수가 계속 증가하는 것과 마찬가지로 우려 사항입니다.,
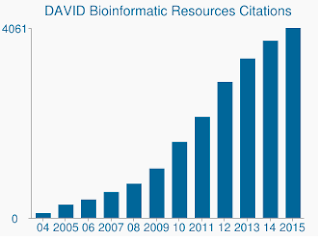
다윗은 아직도의 수천을 받 citations per year 에도 불구하고 기본이 되는 데이터 비참하게 날짜입니다.
결론에는 연구자들은 사용하는 도구에서 보는 전체 프로파일을 결정하는 농축(GSEA,카메라,WilcoxGST,Pathifier)에 복종시키기 보다는 오히려 유전자를 위한 세트를 겹치 분석합니다. 데이비드를 사용하는 것이 빠르고 쉬운 옵션 일지 모르지만,이것은 감도의 엄청난 감소를 희생시키면서 제공됩니다.
추가 읽기
JA Timmons,KJ Szkop,IJ Gallagher., 바이어스의 여러 출처는 글로벌 오믹스 데이터의 기능적 농축 분석을 혼동합니다. 게놈 생물학,2015. 16:186
리나 와디,모나 마이어,조엘 와이저,링컨 디 스타 인,주리 레이망. 경로 농축 분석에 대한 지식 축적의 영향. 바이오 르 시브. doi:http://dx.doi.org/10.1101/049288
편집:2016 년 5 월에 DAVID 가 업데이트되었습니다!
https://david.ncifcrf.gov/content.jsp?file=release.html















