Frontiers in Endocrinology (Português)
Introduction
The tumoral suppressor gene TP53 has been the most heavily studied human gene since its discovery nearly 40 years ago (1). A principal razão por trás deste status é o papel crítico p53 desempenha na prevenção do desenvolvimento do câncer, e é amplamente considerado como o “guardião do genoma”.,”Por algum tempo tem sido geralmente acreditado que o papel de p53 na supressão de tumor é em virtude de sua capacidade de induzir a apoptose, prisão do ciclo celular e senescência de células pré-cancerosas (2). No entanto, é cada vez mais claro que o p53 regula muitas outras vias na célula e que essas outras vias também desempenham papéis na capacidade de p53 funcionar como um supressor de tumor (3). Em particular, o papel da p53 na regulação dos genes envolvidos no metabolismo e na ferroptose tem sido implicado na sua capacidade de suprimir o desenvolvimento do tumor., A ferroptose é uma nova via de morte celular caracterizada pela primeira vez em 2012 e pode ser melhor descrita como uma forma de morte celular dependente do ferro, independente da caspase, impulsionada pela formação de peroxidação lipídica (4). Especificamente, dois modelos de ratos contendo mutações de engenharia em p53 que eliminam a capacidade de P53 para induzir apoptose e senescência ambos retêm a capacidade de suprimir o desenvolvimento espontâneo de tumor; ambos os mutantes retêm a capacidade de transativar genes no metabolismo e ferroptose (5, 6)., Segue-se um resumo dos dados que implicam a p53 na regulação do metabolismo e da ferroptose.
Wild-Type (WT) p53 Positivamente Regula a Fosforilação Oxidativa e Suprime o Metabolismo da Glicose
tipo Selvagem p53 regula a versatilidade metabólica das células, favorecendo a respiração mitocondrial durante a glicólise, em parte, através da transativação da SCO2 (citocromo c oxidase assembly), que desempenha um papel direto na fosforilação oxidativa (7)., a p53 também regula diretamente a transativação da GLS2 (Glutaminase 2); esta enzima permite o uso da glutamina como fonte de energia para as mitocôndrias (8). Além disso, A WT p53 regula negativamente a glicólise, reprimindo transcritivamente os transportadores de glucose GLUT1 e GLUT4, e transativando RRAD e TIGAR; ambos são inibidores da glicólise (9-11). Finalmente, a p53 também se liga e inibe directamente a enzima glucose-6-fosfato desidrogenase, suprimindo assim o metabolismo da glucose (12)., É evidente a partir destes e de outros estudos que, em organismos normais e não comprimidos, o p53 regula directamente o estado metabólico de uma célula (Figura 1). Não surpreendentemente, este gene e muitos dos seus reguladores estão implicados em doenças metabólicas, incluindo obesidade e diabetes (13).
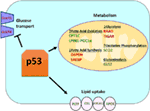
a Figura 1. O papel de tipo selvagem (WT) p53 no metabolismo. Genes regulados positivamente por p53 são mostrados em verde, e genes regulados negativamente por p53 são mostrados em vermelho., o p53 inibe o transporte de glucose, a glicólise e a síntese de ácidos gordos, enquanto promove a captação de lípidos, a oxidação de ácidos gordos, a fosforilação oxidativa e a glutaminólise.
p53 Mutante Positivamente Regula Warburg Metabolismo (Glicólise Aeróbica)
Em contraste com a função de WT p53 mutante do gene p53 em células tumorais favorece a glicólise aeróbica, em parte, reforçando o tráfico de glucose transporter GLUT1 para a membrana plasmática, aumentando, assim, a glicose de importação (14, 15)., Após a mutação de p53, os níveis reduzidos de SCO2 e GLS2 e os níveis aumentados de GLUT1 e GLUT4 favorecem a glicólise aeróbica sobre a fosforilação oxidativa. Desta forma, acredita-se que o p53 mutante contribua para a propensão das células tumorais a utilizar glicólise aeróbica em favor da fosforilação oxidativa, ou o chamado metabolismo de Warburg (15). Uma das características do câncer é o metabolismo desregulado, geralmente demonstrado por esta mudança de glicólise aeróbica para fosforilação oxidativa., Embora isto resulte em uma menor e menos eficiente produção de ATP, acredita-se que as células cancerosas se beneficiam desviando intermediários glicolíticos para vias biossintéticas necessárias para a rápida divisão celular (16). Esta mudança metabólica também leva à diminuição da apoptose mediada pela mitocôndria e à sinalização mais eficiente através dos metabolitos disponíveis nas células cancerígenas (17).
uma variante genética comum em TP53 influencia a sua função no metabolismo
existe um polimorfismo de região de codificação comum de p53 no codon 72, codificando para prolina (P72) ou arginina (R72)., Esta variação de aminoácidos pode ter impacto na função p53 no que diz respeito ao destino celular após stress. Em resposta à lesão do ADN, a variante P72 da p53 desencadeia predominantemente a paragem do ciclo celular, enquanto a variante R72 induz predominantemente a morte celular, ou apoptose (18, 19). Apesar destas diferenças de função, a variação do codon 72 não tem sido consistentemente associada a susceptibilidade ao cancro (20). Em contraste, em estudos humanos este polimorfismo está significativamente associado ao aumento do Índice de massa corporal e risco para a diabetes (21, 22)., Esta premissa é apoiada por estudos em ratinhos, onde um modelo de ratinho para estas variantes do codon 72 mostra o aumento da diabetes induzida pela dieta em ratinhos com a variante R72, em comparação com P72. Nestes estudos, os genes-alvo P53 TNFa e NPC1L1 foram identificados como reguladores críticos no aumento da obesidade induzida pela dieta em ratinhos R72 (23). Curiosamente, a variante R72 também demonstrou conferir uma maior sobrevivência das células em resposta à privação de nutrientes (24)., Estes achados levaram à hipótese de que a variante R72 de p53 surgiu e foi selecionada para como populações migraram para o norte, onde o tempo frio exigiria aumento da acumulação de gordura, mas onde a sobrevivência em resposta à privação de nutrientes também estaria sob seleção (24).
P53 regula o metabolismo lipídico
embora p53 seja bem conhecido por regular a glicólise e o ciclo do ácido cítrico, p53 também tem sido mostrado para desempenhar um papel na regulação do metabolismo lipídico (25)., Acredita-se que WT p53 aumenta a oxidação de ácidos graxos enquanto inibe a síntese de ácidos graxos, agindo assim como um regulador negativo da síntese lipídica (25). Existem vários genes-alvo p53 com papéis no metabolismo lipídico. Sanchez-Macedo e colegas demonstraram que a carnitina palmitoiltransferase 1C (CPT1C) é transcritivamente regulada por p53; esta enzima ajuda no transporte de ácidos gordos ativados para a mitocôndria., Em apoio a um papel para este gene p53 regulado no câncer, este grupo mostrou que ratos deficientes em Cpt1c apresentam desenvolvimento tumoral retardado e taxas de sobrevivência mais elevadas (26). Lipin 1 (LPIN1) é outro gene alvo p53; LPIN1 é necessário para o desenvolvimento adequado de adipócitos e é induzido em condições de baixo nutriente (27). Finck e colegas mostraram que o LPIN1 interage com o PGC-1α, outro gene alvo conhecido do p53 com um papel no metabolismo, e que esta interação ativa a expressão dos genes envolvidos na promoção da oxidação de ácidos graxos (28).,
In addition to directly regulating the transcription of genes involved in lipid metabolism, p53 can also regulate lipid metabolism in a manner involving direct protein-protein interaction. Por exemplo, a glucose-6-fosfato desidrogenase, que é a enzima limitadora da velocidade na Via do fosfato de pentose, liga-se e é directamente inibida pela p53, resultando na diminuição da produção de NADPH e consequentemente na diminuição da síntese de ácidos gordos (12)., The sterol regulatory element-binding proteins (SREBP) family of transcription factors modulate the expression of genes involved in colestrol, fatty acid, triacilglicerol, and phospholipid synthesis (29-31). WT p53 reprime a função SREBP (32), enquanto formas mutantes de p53 se ligam diretamente a SREBP e melhoram sua função transcritional, levando ao aumento da atividade SREBP em tumores humanos (33, 34). Consequentemente, o p53 mutante está correlacionado com uma maior expressão de genes de biossíntese de esterol em tumores mamários humanos (34, 35)., Finalmente, a proteína cinase activada por AMP (AMPK) é uma enzima que é activada sob baixos níveis de nutrientes ou stress energético e é conhecida por inibir a síntese de ácidos gordos interagindo com acetil-CoA-carboxilase e SREBP-1 (36, 37). Zhou e colegas demonstraram que o mutante p53 se liga preferencialmente a AMPK e inibe-o, conduzindo a um aumento da síntese de ácidos gordos. Como resultado, as proteínas p53 mutantes levam ao aumento da sinalização de AMPK, contribuindo para o crescimento celular invasivo das células tumorais (33). A lesser explored area is the role of p53 in lipid transport., It has been shown that p53 transcriptionally regulates apolipoprotein B (apoB) and apoB editing enzyme complex 1, indicating the role of p53 in regulating atherogenic lipoproteins (38). A análise de Microarray de células derivadas do fígado humano identificou a proteína de transferência fosfolípida, a cassete de ligação ATP A12 e a lipase de éster carboxílico como três genes-alvo p53 que todos desempenham um papel no transporte de lípidos (39, 40)., Em geral, embora seja claro que p53 desempenha um papel fundamental na mediação da síntese e metabolismo lipídicos, a contribuição desta via, e estes genes alvo p53, para a supressão tumoral por p53 permanece a ser determinada (figura 1).a Ferroptose é uma nova via de morte celular impulsionada pela peroxidação lipídica
Em 2012, Dixon e colegas descobriram uma nova forma de morte celular regulamentada chamada ferroptose. A ferroptose é uma forma de morte celular dependente do ferro, independente da caspase, resultante da acumulação de lípidos oxidados (4, 41)., Este processo é conduzido pela inactivação da glutationa peroxidase 4 (GPX4), uma enzima que é responsável pela conversão de hidroperóxidos lipídicos letais em álcoois lipídicos não tóxicos, o que requer glutationa para funcionar (41). Acredita-se que a peroxidação dos ácidos gordos poli-insaturados (PUFAs) é o impulso impulsionador para a morte celular por ferroptose. PUFAs contém protões bis-alílicos que podem facilmente ser abstraídos e produzir radicais que irão reagir com oxigênio, criando mais radicais e resultando em uma reação em cadeia de espécies de oxigênio lipídico reativo (42)., O mecanismo exato da morte celular por ferroptose permanece desconhecido, mas uma hipótese é que a lesão lipídica leva à destruição da membrana plasmática (43). Especula-se que a ferroptose pode ser um mecanismo de supressão de tumor que funciona através da eliminação de células que são desprovidas de nutrientes ou que foram expostas a um estresse ambiental ou infecção.,
Farmacológico Regulamento de Ferroptosis
Ferroptosis pode ser induzida pelo uso de inibidores do sistema xc− como erastin, ou análogos, tais como o glutamato e sorafenib, que inibem a importação de cistina, resultando em menos de glutationa e posterior inativação da GPX4. Alternativamente, a ferroptose pode ser induzida por (1S, 3R) – RSL3 (a seguir referido como RSL3), que se liga e inibe diretamente a GPX4 (4, 5, 42). Butiona sulfoximina, FIN56, FINO2, CCl4 e cisplatina são outros agentes que têm sido demonstrados para induzir ferroptose nas células., A morte por ferroptosis pode ser prevenida pela supressão da peroxidação lipídica, o que pode ser feito usando antioxidantes lipofílicos, tais como ferrostatin-1, liproxstatin-1, ou vitamina E. quelantes de Ferro, como a deferoxamina ou cicloprox são outra ferramenta usada para suprimir ferroptosis reduzindo os níveis de ferro. Esgotar PUFAs ou adicionar ácidos gordos monoinsaturados aos meios de cultura celular também pode salvar células da ferroptose (42, 44).,
a Ferroptose está implicada na supressão tumoral mediada pela p53
Em 2012, Gu e colegas desenvolveram um modelo de ratinho no qual três resíduos de lisina normalmente acetilados no domínio de ligação ao ADN da p53 foram mutados para a arginina, pelo que não puderam ser acetilados; este ratinho é referido como o ratinho 3KR. Notavelmente, as células do Ratinho 3KR são incapazes de passar por apoptose dependente de p53, prisão do ciclo celular, ou senescência, e na verdade o mutante 3kr de p53 não consegue transativar a maioria dos genes-alvo p53., Curiosamente, este modelo de rato não desenvolve espontaneamente câncer, implicando que p53 poderia suprimir o desenvolvimento do tumor independente da senescência ou apoptose (45). Este grupo concluiu que a proteína 3kr mutante mantém a capacidade de sofrer ferroptose e regular o metabolismo da cistina, regulando a expressão do Importador de cistina SLC7A11; isto sugere que a ferroptose pode ser uma via que está subjacente à supressão de tumor mediada pela p53., Quando os tipos selvagens e os MEF 3KR foram tratados com a Erastina indutora da ferroptose, observou-se quase 50% de morte celular, enquanto que os MEF nulos p53 exibiam 20% de morte celular, o que indica que o p53 sensibiliza as células para a ferroptose, e também que outros reguladores-chave também desempenham um papel na ferroptose (5). Posteriormente, Gu e colegas identificaram um local adicional de acetilação na lisina 98 de p53, e eles geraram um modelo de ratinho no qual todos os quatro locais de acetilação foram mutados para arginina (4KR)., Curiosamente, o mutante 4KR foi incapaz de regular genes envolvidos em ferroptose como SLC7A11, e ao contrário do mutante 3KR foi incapaz de suprimir o desenvolvimento do tumor (46). Embora atualmente correlativo, estes dados implicam o papel de p53 na ferroptose em sua capacidade de suprimir o desenvolvimento do tumor.
em células não transformadas, p53 regula positivamente a Ferroptose
além do SLC7A11, vários outros genes-alvo directos p53 foram descobertos para desempenhar um papel na ferroptose. Estes incluem GLS2, PTGS2 e SAT1., Estudos de dois grupos separados apoiam o papel do GLS2 na ferroptose, que é conhecido por diminuir a glutationa e aumentar os níveis celulares ROS. Jiang e colegas usaram inibidores da ferroptose combinados com inibidores da glutaminólise para inibir a ferroptose induzida pela Erastina, demonstrando assim que a ferroptose requer glutaminólise e GLS2 (47). Murphy e colegas mostraram que uma variante polimórfica de p53 foi capaz de induzir a paragem do crescimento e a senescência tanto nas células humanas como nas células murinas, mas não conseguiu reprimir SLC7A11 ou transativar o GLS2., Esta variante foi significativamente prejudicada na indução de ferroptose e na supressão do desenvolvimento tumoral, implicando novamente o papel da p53 na supressão tumoral mediada por ferroptose (48). Outro gene alvo p53 com um papel na ferroptose é PTGS2, um gene que codifica a enzima ciclo-oxigenase-2. Stockwell e colegas mostraram pela primeira vez que a indução de ferroptose usando Erasina e RSL3 levou à upregulação de PTGS2 (41). Notavelmente, PTGS2 não foi upregulado por indutores de ferroptose em células nulas p53, sugerindo que este regulamento é dependente de p53 (5)., Atualmente, o upregulation do PTGS2 é amplamente usado como um marcador de ferroptose (5, 41).
Um estudo recente do Grupo Gu mostrou que o gene alvo P53 SAT1 regula a ferroptose (49). Os autores identificaram SAT1 como um alvo direto de p53 e mostraram que o silenciamento de SAT1 reduziu a morte celular induzida por espécies reativas de oxigênio em células com WT p53, mas não teve efeito em células p53-nulas. Mecanicamente, este grupo mostrou que o SAT1 aumenta o nível e a actividade do aracidonato 15-lipoxigenase, uma enzima de ligação ao ferro que oxida os PUFAs e aumenta a peroxidação lipídica., Notavelmente, este estudo mostrou que nem p53 nem SAT1 isoladamente parecem ser suficientes para induzir ferroptose. Em vez disso, os dados combinados são mais consistentes com a premissa de que p53, em virtude de regular genes que contribuem para a ferroptose, regula a sensibilidade das células a esta via, em vez de induzir diretamente ferroptose. Falta determinar se o p53 regula outros genes envolvidos na ferroptose (Figura 2).
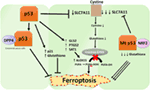
Figura 2. The various roles of p53 in ferroptosis., Inibição da glutationa peroxidase 4( GPX4), a enzima chave que cataliza a conversão de ácidos gordos poli-insaturados (PUFAs) contendo peróxidos em álcoois, é o principal motor da ferroptose. Dependendo do contexto, p53 pode suprimir a ferroptose (como nas células do câncer colorectal) ou promover a ferroptose. O p53 mutante sensibiliza as células para a ferroptose ainda mais do que o tipo selvagem p53.,
em algumas células, p53 regula negativamente a Ferroptose
um estudo recentemente publicado por Tarangelo e colegas mostra que p53 regula negativamente a ferroptose em células cancerígenas (50). Este grupo descobriu que o pré-tratamento de células com Nutlin-3, um composto que estabiliza p53 atrasa o início da ferroptose em vários tipos de células. O início tardio da ferroptose foi encontrado para depender do CDKN1A (codificação p21), um alvo crítico de transcrição p53., O mecanismo através do qual p21 atrasa a ferroptose ainda não foi elucidado, mas acredita-se que a conservação da glutationa intracelular pode ser um fator que contribui para a redução da sensibilidade à ferroptose. Os autores concluem que o eixo p53–p21 permite que as células cancerígenas sobrevivam em condições de estresse metabólico, como privação de cistina, suprimindo o início da ferroptose (50). Um estudo recente mostrou que a p53 inibe a ferroptose nas células cancerígenas colorectais ligando-se à enzima dipeptidil-peptidase-4 (DPP4), que é um modulador da ferroptose e metabolismo lipídico., Mecanicamente, este estudo mostrou que o p53 antagoniza a ferroptose sequestrando o DPP4 em um pool Nuclear enzimático inativo. Na ausência de p53, DPP4 é livre para interagir e formar um complexo com NOX1; isso leva a aumento da peroxidação lipídica e ferroptose. A inibição da DPP4 suprime significativamente a ferroptose, enquanto que a sobreexpressão da DPP4 despoleta a sensibilidade à Erastina, particularmente nas células com depleção de p53 (51). O controlo bidirecional da ferroptose por p53 através de mecanismos dependentes da transcrição e independentes da transcrição pode ser dependente do contexto ou do tipo celular (Figura 2).,
o polimorfismo P47S do TP53 afeta a Ferroptose e a supressão tumoral
além das mutações missense, existem vários polimorfismos de um nucleótido único funcionalmente significantes (SNPs) no gene TP53 e outras proteínas conhecidas para regular esta via (tais como MDM2 e MDM4). A variante Pro47Ser (a seguir designada S47) é a segunda variante mais comum do SNP encontrada na região de codificação p53 (após Pro72Arg) que altera a sequência de aminoácidos da proteína., Para melhor elucidar o impacto desta variante sobre a função p53 e o risco de câncer, o Grupo Murphy gerou um modelo humanizado p53 knock-in mouse, no qual exons 4-9 de murine p53 foram substituídos por exons humanos p53 contendo o tipo selvagem ou a variante S47 (52-55). A maioria dos ratos S47 desenvolveu espontaneamente tumores de vários tipos histológicos, particularmente cancro do fígado, entre 12 e 18 meses de idade, ao contrário dos ratos WT p53 (48)., Nos fibroblastos embrionários do Ratinho e nas linhas celulares linfoblastóides humanas, a variante S47 mostrou diminuição da morte celular programada em resposta à cisplatina e a outras tensões genotóxicas. Mecanicamente, a variante S47 é defeituosa para a transativação de genes envolvidos no metabolismo, tais como Gls2 (glutaminase 2) e Sco2 (48). Consistente com o papel do Gls2 na ferroptose, este grupo descobriu que as células S47 eram marcadamente resistentes aos agentes indutores da ferroptose, Erasina e RSL3 (47, 48). Este defeito pode contribuir para o fenótipo propenso ao tumor observado em ratos S47.,p53 mutante sensibiliza as células tumorais para a Ferroptose P53 tipo selvagem regula negativamente a expressão do Importador de cistina SLC7A11, que inibe a sensibilidade à ferroptose (5). Embora esta regulação ocorra em células normais, em células tumorais, outros mediadores de SLC7A11 parecem predominar na regulação deste gene. Por exemplo, o Fator de transcrição principal do antioxidante NRF2 também pode regular a expressão do SLC7A11 no nível de transcrição, e o NRF2 tem sido implicado como um jogador chave na proteção das células cancerosas contra a ferroptose., Por exemplo, a inibição do NRF2 nas células cancerígenas hepatocelulares aumenta a actividade anti-cancerígena do Erasstin e do Sorafenib in vivo (56). Formas mutantes de p53 podem inibir a função NRF2 por interação direta, e um grupo descobriu que tumores com p53 mutante contêm níveis muito baixos de SLC7A11, e, assim, mostram aumento da sensibilidade à ferroptose. Notavelmente, a sobreexpressão do SLC7A11 em modelos p53 mutantes levou à resistência ao fármaco, sugerindo que os níveis de expressão do SLC7A11 devem ser considerados ao atingir cancros mutantes impulsionados pelo p53 com compostos indutores de ferroptose (57)., Em apoio a esta premissa, o trabalho recente no câncer colorectal (CRC), onde a mutação ou exclusão de p53 é um evento frequente, mostrou que as linhas celulares CRC humanas que abrigam o mutante p53 eram muito mais sensíveis à morte celular mediada pela Erastina quando comparado com as células CRC com WT p53. Para validar estes achados, eles mostraram que knock em uma mutação p53 hotspot em ambas as células HCT116 e SW48 restaurou a sensibilidade à Erastina (51). Estes dados destacam um novo mecanismo pelo qual os cancros impulsionados pelo mutante p53 pode ser explorado usando terapia orientada.,
conclusão
o papel da p53 no metabolismo é bastante claro e possivelmente até intuitivamente óbvio: A WT p53 limita o metabolismo da glucose e a síntese lipídica, enquanto a mutante p53 parece fazer o oposto. A contribuição de seu papel metabólico para a supressão de tumor por p53, e para a capacidade do mutante p53 para conduzir a progressão do tumor, continua a ser inequivocamente provado. O papel do p53 na regulação da ferroptose, e a contribuição desta função, para a supressão do tumor é ainda menos claro., Enquanto dados convincentes de modelos de mouse suportam a premissa de que p53 regula a sensibilidade das células à ferroptose, isso pode ser restringido à capacidade da basal p53 para suprimir o desenvolvimento espontâneo de tumor, e em modelos de mouse com estresse de oncogeno, é claro que a senescência e apoptose desempenham o papel predominante. Da mesma forma, p53 pode regular a sensibilidade à ferroptose de uma forma específica do tipo celular. Mais estudos em modelos animais, com atenção à ferroptose em diferentes tecidos, precisa ser feito para compreender melhor o papel da p53 na ferroptose e ferroptose na supressão de tumor., Além disso, uma ideia mais clara do que os genes-alvo p53 desempenham um papel na sensibilidade à ferroptose precisa ser alcançada. A resolução destas questões deve proporcionar novas vias para combater os tumores com o mutante p53.
contribuições dos autores
KG, SB, TB, AB-K, C-PK, e MM cadA um escreveu um a dois parágrafos deste artigo. KG e SB fizeram a figura. KG e MM delinearam o capítulo.,
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
O revisor OAF e o editor de manuseio declararam sua afiliação compartilhada.
Agradecimentos
a Pesquisa relatada nesta publicação foi apoiada pelos Institutos Nacionais de Saúde, nos termos do Prémio Números CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK), e T32 CA009171 (TB)., O conteúdo é da exclusiva responsabilidade dos autores e não representa necessariamente as opiniões oficiais dos Institutos Nacionais de saúde.
3. Humpton TJ, Vousden KH. Regulação do metabolismo celular e hipoxia por p53. Cold Spring Harb Perspect Med (2016) 6(7):211-30. doi: 10.1101/cshperspect.a026146
CrossRef Texto Completo | Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., O supressor do Tumor p53 regula negativamente a glicólise estimulada pela hipoxia através do seu RRAD alvo. Oncotarget (2014) 5(14):5535-46. doi: 10.18632 / oncotarget.2137
PubMed Resumo | CrossRef Texto Completo | Google Scholar
17. Lee M, Yoon JH. Interação metabólica entre glicólise e oxidação mitocondrial: o efeito Warburg reverso e sua implicação terapêutica. World J Biol Chem (2015) 6(3):148-61. doi: 10.4331 / wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., A depleção do novo gene p53-alvo carnitina palmitoiltransferase 1C atrasa o crescimento do tumor no modelo do tumor de neurofibromatose tipo I. A Morte Celular É Diferente (2013) 20(4):659-68. doi: 10.1038 / cdd.2012.168
PubMed Resumo | CrossRef Texto Completo | Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Disregulação de proteínas de ligação do elemento de resposta do esterol e efectores a jusante no cancro da próstata durante a progressão para a independência androgénica., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA Daming agents: a new biomedical research tool. Oncogeno (2001) 20(3):320-8. doi: 10.1038 / sj.onc.1204080
PubMed Resumo | CrossRef Texto Completo | Google Scholar
57. Liu DS, Duong CP, Haupt s, Montgomery KG, House CM, Azar WJ, et al. A inibição do sistema do eixo xC-/glutationa Visa selectivamente cancros com acumulação mutante-p53., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















