mancha do genoma
análise da Via tornou-se um procedimento comum na Bioinformática, especialmente no estudo da expressão do gene. Se você olhar para uma pesquisa de jornais recentes, parece que há um monte de maneiras que pode ser feito. Neste post, vou discutir as diferenças entre duas ferramentas comumente usadas: DAVID e GSEA.

Os conceitos por trás dos dois algoritmos são muito diferentes., DAVID determina sobreposições entre listas de genes fornecidas pelo Usuário e as bases de dados curadas, procurando sobreposições que são maiores do que o esperado por acaso Aleatório. Você pode melhorar a precisão do algoritmo fornecendo um arquivo de fundo que contém todos os genes que foram considerados/detectados no experimento. Os conjuntos de genes fornecidos pelo usuário são normalmente gerados pela seleção de genes que passam por um limiar de significância. O procedimento DAVID é semelhante a outros disponíveis, tais como ingenuidade, AmiGO e GeneGO., A seleção de valores significativos é em grande parte arbitrária, mas é comum definir o limiar em FDR ajustado p<0,05. A alteração do limiar alterará consideravelmente os resultados do enriquecimento. Os testes estatísticos utilizados incluem hipergeométrico, exato de Fisher e Qui-quadrado.
Em contraste com os métodos que requerem a lista de genes que passam arbitrário limites, GSEA é uma ferramenta que utiliza cada ponto de dados em estatística algoritmo. No método” clássico ” os genes são classificados pela maioria dos up-regulated para a maioria down-regulated., A métrica de classificação em si varia, mas dois métodos válidos são usar o valor p assinado, ou menor intervalo de confiança de 90% da mudança de dobra. A base do teste é avaliar se os membros de um conjunto genético parecem enriquecidos numa extremidade do perfil. Para testar o enriquecimento, a GSEA efectua permutações do perfil, calculando o enriquecimento do gene umas mil ou mais vezes para estimar os valores p empiricamente. Outras ferramentas que usam todo o perfil realizam testes analíticos como o teste de Wilcoxon e Kolmogorov-Smirnov., agora que entendemos as principais diferenças nos algoritmos, podemos começar a comparar os resultados dessas duas técnicas. Revisitei um conjunto de dados que discuti antes, o tratamento com azacitidina em células AML3 perfilado por RNA-seq. Usarei os dados de expressão diferencial RNA-seq gerados pelo DESeq como ponto de partida para comparar DAVID e GSEA.para DAVID, eu fiz um arquivo de fundo consistindo de todos os 19.030 genes detectados no experimento acima do limiar de detecção. O significativo (FDR<0.,05) listas de genes na direção up (2502 genes) e down (2907 genes) foram submetidas separadamente a DAVID 6.7 para análise, focando apenas as vias KEGG.
Para GSEA, eu usei o mesmo procedimento métrico rank descrito anteriormente. Usei o algoritmo clássico de GSEA com o gsea2-2.2.2.executável do jar, usando conjuntos de genes do KEGG do MSigDB v5. 1.
no gráfico abaixo, i determinou a sobreposição de conjuntos de genes estatisticamente significantes (FDR<0,05) separadamente para vias regulamentadas para cima e para baixo., Eu também incluí uma intersecção dos resultados de DAVID quando nenhum fundo é fornecido (apenas por curiosidade, não um procedimento recomendado).
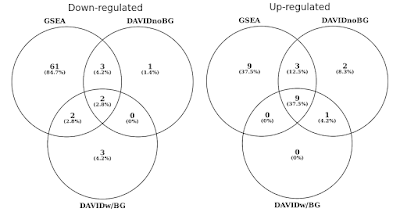
DAVID vs GSEA usando o mesmo perfil de expressão genética. Os números representam conjuntos de genes que são significativamente enriquecidos (FDR<0,05).
você vai notar que tanto na direção para cima e para baixo regulada, GSEA identifica resultados mais significativos em comparação com DAVID. Eu descobri que este também era o caso em outras bibliotecas de conjuntos de genes, como REACTOME e GO., A diferença na sensibilidade entre as duas ferramentas resume-se aos diferentes algoritmos, com DAVID usando uma lista discreta de genes, versus GSEA que usa cada ponto de dados. GSEA será muito mais sensível na identificação de conjuntos de genes que têm muitos membros que estão passando por mudanças sutis na expressão. Individualmente, estas alterações não são estatisticamente significativas, mas quando uma grande fração de genes em um conjunto coletivamente mudança na expressão, essa tendência pode ser muito significativa estatisticamente falando.,in addition to the more sensitive algorithm, DAVID’s gene sets and gene annotations are really out of date. A última atualização das muitas fontes de dados de DAVID está em meados do final de 2009. Como tal, muitas adesões de genes de Ensembl pode não ser suportado. De fato, a partir do meu arquivo de antecedentes que continha 19.030 apenas 13.488 foram reconhecidos por DAVID. A falta de conjuntos de genes atualizados é também uma preocupação, como é o número crescente de citações que DAVID ainda recebe.,
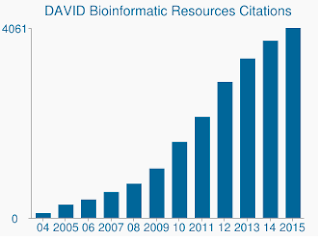
DAVID ainda recebe milhares de citações por ano, apesar dos dados subjacentes estarem lamentavelmente desactualizados.
Em conclusão, eu recomendaria aos pesquisadores usar ferramentas que olham para um perfil inteiro para determinar o enriquecimento (GSEA, CAMERA, WilcoxGST, Pathifier) ao invés de submeter conjuntos de genes para análise sobreposta. Usar DAVID pode ser a opção rápida e fácil, mas isso vem à custa de uma redução massiva na sensibilidade.JA Timmons, KJ Szkop, IJ Gallagher., Múltiplas fontes de viés confundem a análise de enriquecimento funcional de dados globais-omics. Genoma biology, 2015. 16: 186 Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Impacto da acumulação de conhecimentos na análise do enriquecimento das vias. bioRxiv. DOI: http://dx.doi.org/10.1101/049288
EDIT: In May 2016, DAVID was updated!
https://david.ncifcrf.gov/content.jsp?file=release.html















