Genome Spot
Pathway analysis har blivit ett så vanligt förfarande inom bioinformatik, särskilt vid studier av genuttryck. Om man tittar på en undersökning av de senaste tidningarna verkar det som om det finns en massa sätt att det kan göras. I det här inlägget ska jag diskutera skillnaderna mellan två vanliga verktyg; DAVID och gsea.

begreppen bakom de två algoritmerna är väldigt olika., DAVID bestämmer överlappningar mellan användartillförda genlistor och de kurerade databaserna och letar efter överlappningar som är större än vad som förväntas av slumpmässig chans. Du kan förbättra noggrannheten i algoritmen genom att tillhandahålla en bakgrundsfil som innehåller alla gener som ansågs / upptäckts i experimentet. De användartillförda genuppsättningarna genereras normalt genom att välja gener som passerar en signifikansgräns. DAVID förfarandet liknar andra tillgängliga såsom Uppfinningsrikedom, AmiGO och GeneGO., Valet av signifikansvärden är till stor del godtyckligt, men det är vanligt att ställa in tröskeln vid FDR justerade p< 0,05. Om tröskelvärdet ändras kommer anrikningsresultaten att ändras avsevärt. De statistiska tester som används inkluderar hypergeometrisk, Fishers exakta och Chi-squared.
i motsats till ovanstående metoder som kräver listor över gener som passerar godtyckliga tröskelvärden är GSEA ett verktyg som använder varje datapunkt i sin statistiska algoritm. I den” klassiska ” metoden rankas gener av från de flesta uppreglerade till de flesta nedreglerade., Rankmätvärdet i sig varierar, men två giltiga metoder är att använda signerat p-värde eller lägre 90% konfidensintervall för vikändringen. Grunden för testet är att bedöma om medlemmar av en genuppsättning verkar berikade i ena änden av profilen. För att testa anrikningen utför GSEA permutationer av profilen och beräknar anrikningen av genen som en tusen eller flera gånger för att uppskatta p-värden empiriskt. Andra verktyg som använder hela profilen utför analytiska tester som Wilcoxon och Kolmogorov-Smirnov-testet.,
så nu när vi förstår de viktigaste skillnaderna i algoritmerna kan vi börja jämföra resultaten från dessa två tekniker. Jag revisited en dataset jag har diskuterat tidigare, azacitidin behandling i AML3 celler profilerade av RNA-seq. Jag kommer att använda differential-RNA-seq uttryck data som genereras av DESeq som en startpunkt för att jämföra DAVID och GSEA.
för DAVID gjorde jag en bakgrundsfil bestående av alla 19,030-gener som detekterades i experimentet över detektionströskeln. Den signifikanta (FDR< 0.,05) genlistor i upp (2502 gener) och ner (2907 gener) riktning lämnades separat till DAVID 6.7 för analys, med fokus på KEGG vägar endast.
för gsea använde jag samma mätmetod för rankning som beskrivits tidigare. Jag använde den klassiska gsea-algoritmen med gsea2-2.2.2.jar körbar, med KEGG genuppsättningar från MSigDB v5. 1.
i diagrammet nedan bestämde jag överlappningen av statistiskt signifikanta genuppsättningar (FDR<0.05) separat för upp och ner reglerade vägar., Jag inkluderade också en korsning av DAVID resultat när ingen bakgrund tillhandahålls (endast för nyfikenhet, inte ett rekommenderat förfarande).
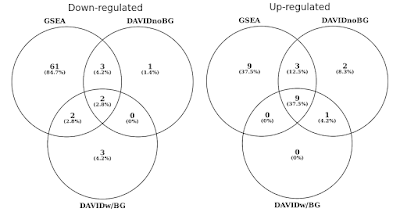
DAVID vs gsea använder samma genuttryck profil. Siffror representerar genuppsättningar som är signifikant berikade (FDR<0.05).
Du kommer att märka att GSEA i både upp och ner reglerad riktning identifierar mer signifikanta resultat jämfört med DAVID. Jag fann att detta också var fallet i andra genuppsättningsbibliotek som REACTOME och GO., Skillnaden i känsligheten mellan de två verktygen kokar ner till de olika algoritmerna, med DAVID som använder en diskret lista över gener, jämfört med gsea som använder varje datapunkt. GSEA kommer att vara mycket känsligare för att identifiera genuppsättningar som har många medlemmar som genomgår subtila förändringar i uttryck. Individuellt är dessa förändringar inte statistiskt signifikanta, men när en stor del av generna i en uppsättning kollektivt skiftar i uttryck, kan den trenden vara mycket signifikant statistiskt sett.,
förutom den känsligare algoritmen är Davids genuppsättningar och genanteckningar verkligen föråldrade. Den senaste uppdateringen av Davids många datakällor är i mitten till slutet av 2009. Som sådan kan många gentillgångar från Ensembl inte stödjas. Faktum är att från min bakgrundsfil som innehöll 19,030 erkändes endast 13,488 av DAVID. Bristen på uppdaterade genuppsättningar är också ett problem, liksom det ständigt växande antalet citat som DAVID fortfarande får.,
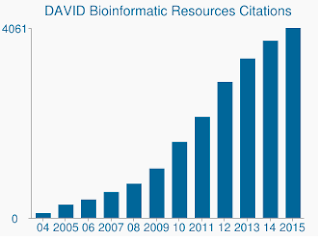
DAVID får fortfarande tusentals citat per år trots att underliggande data är otroligt föråldrade.
Sammanfattningsvis rekommenderar jag forskare att använda verktyg som tittar på en hel profil för att bestämma anrikning (GSEA, CAMERA, WilcoxGST, Pathifier) snarare än att skicka genuppsättningar för överlappningsanalys. Att använda DAVID kan vara det snabba och enkla alternativet, men detta kommer på bekostnad av en massiv minskning av känsligheten.
ytterligare läsning
JA Timmons, kj Szkop, ij Gallagher., Flera källor till bias confound funktionell anrikning analys av global-omics data. Genombiologi, 2015. 16:186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Inverkan av kunskapsackumulering på analys av anrikning av vägar. bioRxiv. doi:http://dx.doi.org/10.1101/049288
Redigera: i maj 2016 uppdaterades DAVID!
https://david.ncifcrf.gov/content.jsp?file=release.html















