gränser i endokrinologi
introduktion
tumörsuppressorgenen TP53 har varit den mest studerade mänskliga genen sedan dess upptäckt nästan 40 år sedan (1). Den främsta orsaken till denna status är den kritiska rollen p53 spelar för att förebygga cancerutveckling, och det anses allmänt som ”väktare av genomet.,”Under en tid har det allmänt trott att p53: s roll i tumörundertryckning är på grund av dess förmåga att inducera apoptos, cellcykelstopp och senescens av pre-cancerceller (2). Det är emellertid nu allt tydligare att p53 reglerar många andra vägar i cellen och att dessa andra vägar också spelar roller i p53: s förmåga att fungera som en tumörsuppressor (3). I synnerhet har p53: s roll i regleringen av gener som är involverade i metabolism och ferroptosis varit inblandad i dess förmåga att undertrycka tumörutveckling., Ferroptosis är en ny celldödsväg som först karakteriseras i 2012 och kan bäst beskrivas som en järnberoende, caspasoberoende form av celldöd som drivs av bildandet av lipidperoxidering (4). Specifikt behåller två musmodeller som innehåller konstruerade mutationer i p53 som eliminerar p53: s förmåga att inducera apoptos och senescence både förmågan att undertrycka spontan tumörutveckling; båda dessa mutanter behåller förmågan att transaktivera gener i metabolism och ferroptosis (5, 6)., En sammanfattning av de data som påverkar p53 i regleringen av metabolism och ferroptosis beskrivs nedan.
Wild-Type (WT) p53 reglerar positivt oxidativ fosforylering och undertrycker glukosmetabolism
Wild-type p53 reglerar cellernas metaboliska mångsidighet genom att gynna mitokondriell andning över glykolys, delvis via transaktivering av SCO2 (cytokrom C-oxidasmontering), som spelar en direkt roll i oxidativ fosforylering (7)., p53 reglerar också direkt transaktiveringen av GLS2 (Glutaminas 2); detta enzym tillåter glutaminanvändning som en energikälla för mitokondrierna (8). Dessutom reglerar WT p53 glykolys negativt genom att transkriptionellt undertrycka glukostransportörerna GLUT1 och GLUT4 och genom att transaktivera RRAD och TIGAR; båda är hämmare av glykolys (9-11). Slutligen binder p53 också direkt och hämmar enzymet glukos-6-fosfatdehydrogenas, vilket undertrycker glukosmetabolism (12)., Det framgår av dessa och andra studier att i normala, ostressade organismer reglerar p53 direkt det metaboliska tillståndet i en cell (Figur 1). Inte överraskande är denna gen och många av dess regulatorer inblandade i metaboliska sjukdomar, inklusive fetma och diabetes (13).
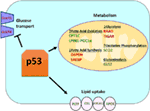
Figur 1. Rollen av vildtyp (WT) p53 i ämnesomsättningen. Gener som positivt regleras av p53 visas i grönt, och gener som negativt regleras av p53 visas i rött., p53 hämmar glukostransport, glykolys och fettsyrasyntes medan det främjar lipidupptag, fettsyraoxidation, oxidativ fosforylering och glutaminolys.
Mutant p53 reglerar positivt Warburg Metabolism (aerob glykolys)
i motsats till funktionen av WT p53, mutant p53 i tumörceller gynnar aerob glykolys, delvis genom att öka handeln av glukostransportören GLUT1 till plasmamembranet, vilket ökar glukosimport (14, 15)., Efter mutationen av p53 gynnar de reducerade nivåerna av SCO2 och GLS2 och de ökade nivåerna av GLUT1 och GLUT4 aerob glykolys över oxidativ fosforylering. På detta sätt tros mutant p53 bidra till benägenheten hos tumörceller att utnyttja aerob glykolys till förmån för oxidativ fosforylering eller så kallad Warburg-metabolism (15). Ett av kännetecknen för cancer är avreglerad metabolism, vilket i allmänhet demonstreras genom denna övergång från aerob glykolys till oxidativ fosforylering., Även om detta resulterar i ett lägre och mindre effektivt ATP-utbyte, antas det att cancerceller gynnas genom att avleda glykolytiska mellanprodukter till biosyntetiska vägar som är nödvändiga för snabb celldelning (16). Denna metaboliska omkopplare leder också till minskad mitokondriermedierad apoptos och effektivare signalering genom tillgängliga metaboliter i cancerceller (17).
en vanlig genetisk Variant i TP53 påverkar dess funktion i ämnesomsättningen
det finns en gemensam kodningsregion polymorfism av p53 vid kodon 72, kodning för antingen prolin (P72) eller arginin (R72)., Denna aminosyravariation kan påverka p53-funktionen när det gäller cellöde efter stress. Som svar på DNA-skada utlöser P72-varianten av p53 övervägande cellcykelstopp, medan R72-varianten huvudsakligen inducerar celldöd eller apoptos (18, 19). Trots dessa skillnader i funktion har kodon 72-variationen inte konsekvent associerats med cancerkänslighet (20). Däremot är denna polymorfism i humana studier signifikant associerad med ökat kroppsmassindex och risk för diabetes (21, 22)., Denna premiss stöds av studier på möss, där en musmodell för dessa kodon 72-varianter visar ökad fetthaltig dietinducerad diabetes hos möss med R72-varianten jämfört med P72. I dessa studier identifierades p53-målgenerna TNFa och NPC1L1 som kritiska regulatorer i ökningen av dietinducerad fetma hos R72-möss (23). Intressant har R72-varianten också visat sig ge ökad överlevnad av celler som svar på näringsbrist (24)., Dessa resultat har lett till hypotesen att R72-varianten av p53 uppstod och valdes för som populationer migrerade norrut, där kallt väder skulle kräva ökad fettackumulering, men där överlevnad som svar på näringsbrist också skulle vara under urval (24).
p53 reglerar lipidmetabolism
även om p53 är välkänt för att reglera glykolys och citronsyracykeln, har p53 också visat sig spela en roll för att reglera lipidmetabolism (25)., Man tror att WT p53 förbättrar fettsyraoxidation samtidigt hämma fettsyrasyntesen, vilket sålunda fungerar som en negativ regulator av lipidsyntes (25). Det finns flera p53 målgener med roller i lipidmetabolism. Sanchez-Macedo och kollegor visade att karnitin palmitoyltransferase 1C (CPT1C) är transcriptionally regleras av p53; detta enzym hjälpmedel vid transport av aktiverade fettsyror till mitokondrier., Till stöd för en roll för denna p53-reglerade gen i cancer visade denna grupp att Cpt1c-bristfälliga möss visar fördröjd tumörutveckling och högre överlevnadshastigheter (26). Lipin 1 (LPIN1) är en annan p53-målgen; LPIN1 är nödvändig för korrekt adipocytutveckling och induceras under låga näringsförhållanden (27). Finck och kollegor visade att LPIN1 interagerar med PGC-1α, en annan känd p53-målgen med en roll i ämnesomsättningen, och att denna interaktion aktiverar uttrycket av gener som är involverade i att främja fettsyraoxidation (28).,
förutom att direkt reglera transkriptionen av gener som är involverade i lipidmetabolism kan p53 också reglera lipidmetabolism på ett sätt som involverar direkt protein–proteininteraktion. Till exempel binder glukos-6-fosfatdehydrogenas, vilket är det hastighetsbegränsande enzymet i pentosfosfatvägen, till och hämmas direkt av p53, vilket resulterar i minskad NADPH-produktion och följaktligen minskad fettsyrasyntes (12)., Sterol regulatory element-bindande proteiner (SREBP) familj av transkriptionsfaktorer modulera uttrycket av gener som är involverade i kolesterol, fettsyra, triacylglycerol, och fosfolipid syntes (29-31). WT p53 undertrycker SREBP-funktionen (32), medan mutanta former av p53 binder direkt till SREBP och förbättrar deras transkriptionella funktion, vilket leder till ökad SREBP-aktivitet i mänskliga tumörer (33, 34). Följaktligen är mutant p53 korrelerad med högre uttryck av sterolbiosyntesgener i humana brösttumörer (34, 35)., Slutligen är AMP-aktiverat proteinkinas (AMPK) ett enzym som aktiveras under låga näringsnivåer eller energistress och är känt för att hämma fettsyrasyntesen genom att interagera med acetyl-CoA-karboxylas och SREBP-1 (36, 37). Zhou och kollegor visade att mutant p53 företrädesvis binder till och hämmar AMPK, vilket leder till ökad fettsyrasyntes. Som ett resultat leder mutant p53-proteiner till ökad AMPK-signalering, vilket bidrar till invasiv celltillväxt av tumörceller (33). Ett mindre utforskat område är rollen som p53 i lipidtransport., Det har visat sig att p53-transkriptionellt reglerar apolipoprotein B (apoB) och apoB-redigeringsenzymkomplex 1, vilket indikerar p53: s roll vid reglering av aterogena lipoproteiner (38). Mikroarrayanalys av humana leverderiverade celler identifierade fosfolipidöverföringsprotein, ATP-bindande kassett A12 och karboxylesterlipas som tre p53-målgener som alla spelar en roll vid lipidtransport (39, 40)., Sammantaget, även om det är uppenbart att p53 spelar en nyckelroll för att mediera lipidsyntes och metabolism, är bidraget från denna väg, och dessa p53-målgener, till tumörundertryckning av p53 fortfarande att bestämmas (Figur 1).
Ferroptosis är en ny Celldödsväg som drivs av lipidperoxidering
under 2012 upptäckte Dixon och kollegor en ny form av reglerad celldöd som heter ferroptosis. Ferroptosis är en järnberoende, caspas-oberoende form av celldöd till följd av ackumulering av oxiderade lipider (4, 41)., Denna process drivs av inaktivering av glutationperoxidas 4 (GPX4), ett enzym som är ansvarigt för att omvandla dödliga lipidhydroperoxider till giftfria lipidalkoholer, vilket kräver glutation för att fungera (41). Man tror att peroxidering av fleromättade fettsyror (PUFAs) är drivkraften för celldöd genom ferroptosis. PUFAs innehåller bis-allylproton som lätt kan abstraheras och producera radikaler som kommer att reagera med syre, vilket skapar fler radikaler och resulterar i en kedjereaktion av lipidreaktiva syrearter (42)., Den exakta mekanismen för celldöd genom ferroptosis är fortfarande okänd, men en hypotes är att lipidskadorna leder till förstörelsen av plasmamembranet (43). Det har spekulerats att ferroptosis kan vara en mekanism för tumörundertryck som fungerar genom att eliminera celler som är näringsämnen berövade eller har utsatts för en miljöbelastning eller infektion.,
farmakologisk reglering av Ferroptosis
Ferroptosis kan induceras med användning av hämmare av system xc – såsom erastin, eller analoger såsom glutamat och sorafenib, som hämmar importen av cystin, vilket resulterar i utarmad glutation och efterföljande inaktivering av GPX4. Alternativt, ferroptosis kan induceras av (1S,3R)-RSL3 (nedan kallad RSL3), som direkt binder till och hämmar GPX4 (4, 5, 42). Buthione sulfoximine, FIN56, FINO2, CCl4, och cisplatin är andra medel som har visat sig inducera ferroptosis i cellerna., Död av ferroptosis kan förebyggas genom att undertrycka lipid peroxidationen, vilket kan åstadkommas med hjälp av fettlösliga antioxidanter, såsom ferrostatin-1, liproxstatin-1, eller vitamin E. Strykjärn kelater som deferoxamin eller cicloprox är ett annat verktyg som används för att undertrycka ferroptosis genom att reducera nivåerna av järn. Nedbrytning av PUFA eller tillsats av enomättade fettsyror till cellkulturmedier kan också rädda celler från ferroptosis (42, 44).,
Ferroptosis är inblandad i p53-medierad Tumöruppression
2012 utvecklade Gu och kollegor en musmodell där tre normalt acetylerade lysinrester i DNA-bindande domänen av p53 muterades till arginin och därför inte kunde acetyleras; denna mus kallas 3KR-musen. I synnerhet kan celler från 3KR-musen inte genomgå p53-beroende apoptos, cellcykelstopp eller senescence, och i själva verket misslyckas 3KR-mutanten av p53 att transaktivera majoriteten av p53-målgener., Intressant är att denna musmodell inte spontant utvecklar cancer, vilket innebär att p53 kan undertrycka tumörutveckling oberoende av senescence eller apoptos (45). Denna grupp fann att det mutanta 3KR-proteinet behåller förmågan att genomgå ferroptosis och reglera cystinmetabolism genom att reglera uttrycket av CYSTINIMPORTÖREN SLC7A11; detta föreslog att ferroptosis kan vara en väg som ligger till grund för p53-medierad tumörundertryckning., När vilda och typ 3KR MEFs behandlades med ferroptosis inducerare Erastin, nästan 50% celldöd observerades medan p53 null MEFs uppvisade 20% celldöd; detta tyder på att p53 mer känsliga celler för att ferroptosis, och även att andra centrala tillsynsmyndigheter som också spela en roll i ferroptosis (5). Därefter identifierade Gu och kollegor ett extra acetyleringsställe vid lysin 98 av p53, och de genererade en musmodell där alla fyra acetyleringsställen muterades till arginin (4KR)., Intressant nog kunde 4kr-mutanten inte reglera gener som var involverade i ferroptosis som SLC7A11, och till skillnad från 3KR-mutanten kunde inte undertrycka tumörutveckling (46). Även om dessa data för närvarande är korrelativa, implicerar dessa data rollen som p53 i ferroptosis i dess förmåga att undertrycka tumörutveckling.
I icke-transformerade celler reglerar p53 positivt Ferroptosis
förutom SLC7A11 har flera andra direkta p53-målgener upptäckts för att spela en roll i ferroptosis. Dessa inkluderar GLS2, PTGS2, och SAT1., Studier från två separata grupper stöder GLS2: s roll vid ferroptosis, vilket är känt för att minska glutation och öka cellulära ROS-nivåer. Jiang och kollegor som används ferroptosis-hämmare i kombination med glutaminolysis-hämmare för att hämma Erastin-inducerad ferroptosis, vilket visar att ferroptosis kräver glutaminolysis och GLS2 (47). Murphy och kollegor visade att en polymorf variant av p53 kunde inducera tillväxtstopp och senescence i både mänskliga och murina celler men misslyckades med att undertrycka SLC7A11 eller transaktivera GLS2., Denna variant försämrades markant vid inducering av ferroptosis och undertryckande tumörutveckling, vilket återigen innebar rollen som p53 i ferroptosmedierad tumörundertryckning (48). En annan p53-målgen med en roll i ferroptosis är PTGS2, en gen som kodar för enzymet cyklooxygenas-2. Stockwell och kollegor visade att introduktionen av ferroptosis med Erastin och RSL3 ledde till uppreglering av PTGS2 (41). Särskilt var PTGS2 inte uppreglerat av ferroptosinducerare i p53-null-celler, vilket tyder på att denna förordning är p53-beroende (5)., För närvarande används upreguleringen av PTGS2 i stor utsträckning som en ferroptosmarkör (5, 41).
En nyligen genomförd studie av Gu-gruppen visade på att p53 målgenen SAT1 reglerar ferroptosis (49). Författarna identifierade SAT1 som ett direkt mål för p53 och visade att ljuddämpning av SAT1 minskade celldöd inducerad av reaktiva syrearter i celler med WT p53, men hade ingen effekt i p53-null-celler. Mekaniskt visade denna grupp att SAT1 ökar nivån och aktiviteten hos arakidonat 15-lipoxygenas, ett järnbindande enzym som oxiderar PUFAs och ökar lipidperoxidering., I synnerhet visade denna studie att varken p53 eller SAT1 ensam verkar vara tillräckliga för att inducera ferroptosis. I stället är de kombinerade uppgifterna mer konsekventa med förutsättningen att p53, genom att reglera gener som bidrar till ferroptosis, reglerar cellernas känslighet för denna väg, snarare än direkt inducerar ferroptosis. Huruvida p53 reglerar andra gener som är involverade i ferroptosis återstår att bestämmas (Figur 2).
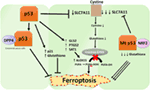
Figur 2. De olika rollerna av p53 i ferroptosis., Hämning av glutationperoxidas 4 (GPX4), det viktigaste enzymet som katalyserar omvandlingen av fleromättade fettsyror (PUFA) innehållande peroxider till alkoholer, är den viktigaste drivkraften för ferroptosis. Beroende på sammanhanget kan p53 undertrycka ferroptosis (som i kolorektala cancerceller) eller främja ferroptosis. Mutant p53 sensibiliserar celler till ferroptosis ännu mer än vildtyp p53.,
i vissa celler reglerar p53 Ferroptosis negativt
en studie som nyligen publicerades av Tarangelo och kollegor visar att p53 reglerar ferroptosis negativt i cancerceller (50). Denna grupp fann att förbehandling av celler med Nutlin-3, en förening som stabiliserar p53 fördröjer uppkomsten av ferroptosis i flera celltyper. Den fördröjda uppkomsten av ferroptosis befanns bero på CDKN1A (kodning p21), ett kritiskt p53-transkriptionellt mål., Mekanismen genom vilken P21 fördröjer ferroptosis har ännu inte klarlagts, men man tror att bevarandet av intracellulär glutation kan vara en bidragande faktor för minskad ferroptoskänslighet. Författarna drar slutsatsen att p53-P21-axeln gör det möjligt för cancerceller att överleva under förhållanden med metabolisk stress, såsom cystinbrist, genom att undertrycka uppkomsten av ferroptosis (50). En nyligen genomförd studie visade att p53 hämmar ferroptosis i kolorektal cancer celler genom att binda till den enzym dipeptidyl-peptidase-4 (DPP4), som är en modulator för ferroptosis och lipidmetabolismen., Mekanistiskt visade denna studie att p53 antagoniserar ferroptosis genom att sekvestrera DPP4 i en nukleär enzymatisk inaktiv pool. I avsaknad av p53 är DPP4 fritt att interagera med och bilda ett komplex med NOX1; detta leder till ökad lipidperoxidering och ferroptosis. Hämning av DPP4 undertrycker ferroptosis signifikant, medan överexpression av DPP4 utlöser Erastin känslighet, särskilt i p53-utarmade celler (51). Dubbelriktad kontroll av ferroptosis genom p53 genom transkriptionsberoende och transkriptionsoberoende mekanismer kan vara kontextberoende eller celltypsberoende (Figur 2).,
P47S Polymorfism av TP53 Påverkar Ferroptosis och Tumör Förtryck
förutom att missense mutationer, det finns flera funktionellt betydande single-nucleotide polymorphisms (Snp) i genen TP53 och andra proteiner som är kända för att reglera denna väg (som MDM2 och MDM4). Pro47ser-varianten (hädanefter S47) är den näst vanligaste SNP som finns i p53-kodningsområdet (efter Pro72Arg) som förändrar proteinets aminosyrasekvens., För att bättre belysa effekten av denna variant på p53-funktion och cancerrisk genererade Murphy-gruppen en humaniserad p53-knock-in-musmodell, där exoner 4-9 av murine p53 ersattes av mänskliga p53-exoner som innehåller antingen den vilda typen eller S47-varianten (52-55). Majoriteten av S47 möss som spontant utvecklade tumörer av olika histologiska typer, särskilt levercancer, mellan 12 och 18 månaders ålder, till skillnad från WT p53-möss (48)., I musembryonala fibroblaster och humana lymfoblastoida cellinjer visade S47-varianten nedsatt programmerad celldöd som svar på cisplatin och andra genotoxiska påfrestningar. Mekaniskt är S47-varianten defekt för transaktivering av gener som är involverade i metabolism, såsom Gls2 (glutaminas 2) och Sco2 (48). Förenligt med rollen som Gls2 i ferroptosis, denna grupp fann att S47 celler var markant resistenta mot ferroptosis-inducerande medel Erastin och RSL3 (47, 48). Denna defekt kan bidra till den tumörbenägna fenotypen som observerats hos S47-möss.,
Mutant p53 sensibiliserar tumörceller till Ferroptosis
Wild-type p53 reglerar negativt uttrycket av cystinimportören SLC7A11, vilket hämmar känsligheten för ferroptosis (5). Även om denna reglering sker i normala celler, verkar andra mediatorer av SLC7A11 dominera i regleringen av denna gen i tumörceller. Till exempel kan Master antioxidant transkription factor NRF2 också reglera uttrycket av SLC7A11 på transkriptionsnivå, och NRF2 har varit inblandad som en nyckelspelare för att skydda cancerceller mot ferroptosis., Till exempel ökar hämning av NRF2 i hepatocellulära cancerceller Anti-canceraktiviteten hos Erastin och Sorafenib in vivo (56). Muterade former av p53 kan hämma NRF2 funktion genom direkt interaktion, och en grupp fann att tumörer med muterat p53 innehåller mycket låga nivåer av SLC7A11, och därmed visa ökad känslighet för ferroptosis. I synnerhet ledde överuttryck av SLC7A11 i mutanta p53-modeller till läkemedelsresistens, vilket tyder på att halter av SLC7A11-uttryck måste beaktas vid inriktning av mutanta p53-drivna cancerformer med ferroptosinducerande föreningar (57)., Till stöd för denna premiss visade det senaste arbetet i kolorektal (CRC) cancer, där mutation eller radering av p53 är en frekvent händelse, att humana CRC-cellinjer som hyser mutant p53 var mycket känsligare för Erastinmedierad celldöd jämfört med CRC-celler med WT p53. För att validera dessa fynd visade de att knackning i en p53-hotspotmutation i både hct116 och SW48-celler återställde känsligheten för Erastin (51). Dessa data belyser en ny mekanism genom vilken cancer som drivs av mutant p53 kan utnyttjas med hjälp av målinriktad terapi.,
slutsats
p53: s roll i ämnesomsättningen är ganska tydlig och kanske till och med intuitivt uppenbar: WT p53 begränsar glukosmetabolism och lipidsyntes, medan mutant p53 verkar göra motsatsen. Bidraget från dess metaboliska roll till tumörundertryck av p53, och till förmågan hos mutant p53 att driva Tumörprogression, återstår att otvetydigt bevisas. Rollen av p53 i regleringen av ferroptosis, och bidraget från denna funktion, till tumörundertryckning är ännu mindre tydlig., Medan övertygande data från musmodeller stöder förutsättningen att p53 reglerar cellernas känslighet för ferroptosis, kan detta begränsas till basal p53: s förmåga att undertrycka spontan tumörutveckling och i onkogen-stressade musmodeller är det uppenbart att senescence och apoptos spelar den dominerande rollen. På samma sätt kan p53 reglera ferroptosen känslighet på ett celltyp-specifikt sätt. Fler studier i djurmodeller, med uppmärksamhet på ferroptosis i olika vävnader, måste göras för att mer fullständigt förstå rollen som p53 i ferroptosis och ferroptosis vid tumörundertryck., Dessutom måste en tydligare uppfattning om vilka p53-målgener som spelar en roll i känsligheten för ferroptosis uppnås. Upplösning av dessa frågor bör ge välbehövliga nya vägar för att bekämpa tumörer med mutant p53.
författare bidrag
KG, SB, TB, AB-K, C-PK, Och MM vardera skrev en till två stycken i denna artikel. KG och SB gjorde figuren. KG och MM beskrivs kapitlet.,
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
granskaren OAF och hanteringsredigeraren förklarade sin delade anslutning.
Tack
Forskning som redovisas i denna publikation stöds av National Institutes of Health under Award Nummer CA102184 (MM), CA201430 (MM), TL1TR002344 (C-PK), och T32 CA009171 (TB)., Innehållet är enbart författarnas ansvar och representerar inte nödvändigtvis de nationella Hälsoinstitutens officiella åsikter.
3. Humpton TJ, Vousden KH. Reglering av cellulär metabolism och hypoxi av p53. Kalla Våren Harb Perspect Med (2016) 6(7):211-30. doi:10.1101/cshperspect.A026146
CrossRef Full Text / Google Scholar
10. Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, et al., Tumörsuppressor p53 reglerar glykolys stimulerad av hypoxi negativt genom sitt mål RRAD. Onkotarget (2014) 5(14):5535-46. doi:10.18632/oncotarget.2137
PubMed Abstract / CrossRef Full Text / Google Scholar
17. Lee M, Yoon JH. Metaboliskt samspel mellan glykolys och mitokondriell oxidation: den omvända Warburg-effekten och dess terapeutiska implikation. World J Biol Chem (2015) 6(3):148-61. doi:10.4331/wjbc.v6.i3.,148
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33(3):357–65. doi:10.1038/ng1093
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME., Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res (2015) 13(2):250–62. doi:10.1158/1541-7786.MCR-14-0385
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab (2011) 24(7–8):437–9. doi:10.1515/jpem.,2011.058
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Kung CP, Liu Q, Murphy ME. The codon 72 polymorphism of p53 influences cell fate following nutrient deprivation. Cancer Biol Ther (2017) 18(7):484–91. doi:10.1080/15384047.2017.1323595
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al., Utarmning av den nya p53-målgenen carnitine palmitoyltransferas 1C fördröjer tumörtillväxt i neurofibromatos typ i tumörmodell. Celldöd Skiljer Sig Åt (2013) 20(4):659-68. doi: 10.1038 / CDD.2012.168
PubMed Abstract / CrossRef Full Text / Google Scholar
30. Ettinger SL, Sobel R, Whitmore TG, Akbari M, DR Bradley, Gleave MIG, et al. Dysregulering av sterol response element-bindande proteiner och nedströms effectors i prostatacancer under progression till androgen oberoende., Cancer Res (2004) 64(6):2212–21. doi:10.1158/0008-5472.CAN-2148-2
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi:10.1073/pnas.,0910258107
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A (2016) 113(34):E4966–75. doi:10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
54., Luo JL, Yang Q, Tong WM, Hergenhahn M, Wang ZQ, Hollstein M. Knock-in möss med en chimerisk människa / murine p53 gen utvecklas normalt och visar vilda typ p53 svar på DNA skadliga ämnen: en ny biomedicinsk forskningsverktyg. Onkogen (2001) 20(3):320-8. doi:10.1038/sj.onk.1204080
PubMed Abstract / CrossRef Full Text / Google Scholar
57. Liu DS, Duong CP, Haupt S, Montgomery KG, Hus CM, Azar WJ, et al. Inhibering av systemet XC – / glutation axel selektivt riktar cancer med mutant – p53 ackumulering., Nat Commun (2017) 8:14844. doi:10.1038/ncomms14844
PubMed Abstract | CrossRef Full Text | Google Scholar















