Genom Spot (Norsk)
Sti analyse har blitt en så vanlig prosedyre i bioinformatikk, spesielt i å studere genuttrykk. Hvis du ser på en undersøkelse av de siste papirene ser det ut til at det er en haug av måter som det kan gjøres. I dette innlegget vil jeg diskutere forskjellene mellom to brukte verktøy; DAVID og GSEA.

konseptene bak de to algoritmene er svært forskjellige., DAVID bestemmer overlapper mellom bruker-leveres genet lister og kuratert databaser, på jakt etter overlapper som er større enn det som er forventet ved tilfeldige. Du kan forbedre nøyaktigheten av algoritmen ved å gi en bakgrunn fil som inneholder alle genene som ble ansett/oppdaget i eksperimentet. Brukeren følger genet normalt sett er generert ved å velge gener som passerer en betydning terskel. DAVID prosedyren er lik andre tilgjengelig som Oppfinnsomhet, AmiGO og GeneGO., Det utvalget av betydning verdier er i stor grad tilfeldig, men det er vanlig å angi grensen på FDR-justerte p<0.05. Endring av terskelen vil endre berikelse resultatene betydelig. Den statistiske tester ansatt inkluderer hypergeometric, Fisher ‘ s eksakte og Chi-squared.
I motsetning til de nevnte metodene som krever lister av gener som passerer vilkårlig terskler, GSEA er et verktøy som bruker hver datapoint i sin statistiske algoritme. I den «klassiske» metode gener er rangert av fra de mest up-regulert til de fleste nedregulert., Rang beregning seg varierer, men to gyldig metodene er å bruke logget p-verdi, eller lavere 90% konfidensintervall på flippen endre. På grunnlag av testen er å vurdere om medlemmer av et gen som set vises anriket i den ene enden av profilen. For å teste berikelse, GSEA utfører permutasjoner av profilen, beregning berikelse av genet sett tusen ganger eller mer for å beregne p-verdier empirisk. Andre verktøy som bruker hele profilen utføre analytiske tester som Wilcoxon og Kolmogorov-Smirnov test.,
Så nå som vi forstår de viktigste forskjellene i algoritmer, kan vi begynne å sammenligne resultatene fra disse to teknikkene. Jeg var på et dataset som jeg har diskutert før, azacitidine behandling i AML3 celler profilert av RNA-seq. Jeg vil bruke differensial-RNA-seq uttrykk data som er generert av DESeq som en start punkt for å sammenligne DAVID og GSEA.
For DAVID, jeg laget en bakgrunn fil som består av alle 19,030 gener oppdaget i eksperimentet over gjenkjennings-terskel. Den betydelige (FDR<0.,05) – genet lister opp (2502 gener) og ned (2907 gener) retning var separat sendt til DAVID 6.7 for analyse, med fokus på KEGG veier bare.
For GSEA, jeg brukte den samme rang beregning prosedyren som er beskrevet tidligere. Jeg brukte den klassiske GSEA algoritmen med gsea2-2.2.2.jar kjørbar, ved hjelp av KEGG genet sett fra MSigDB v5.1.
I grafen under, har jeg bestemt overlapper statistisk signifikant genet sett (FDR<0.05) separat for opp og ned regulert trasé., Jeg har også inkludert et kryss av DAVID resultater når ingen bakgrunn er oppgitt (for nysgjerrighet, og ikke en anbefalt prosedyre).
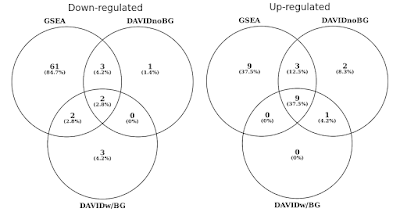
DAVID vs GSEA ved hjelp av det samme genet uttrykk profil. Tallene viser genet sett som er betydelig beriket (FDR<0.05).
Du vil legge merke til at i både opp og ned regulert retning, GSEA identifiserer flere betydelige resultater i forhold til DAVID. Jeg fant dette var også tilfelle i andre gene satt biblioteker som REACTOME og GÅ., Forskjell i følsomhet mellom de to verktøyene koker ned til de ulike algoritmer, med DAVID ved hjelp av en atskilt liste av gener versus GSEA som bruker hvert datapunkt. GSEA vil være mye mer sensitive i å identifisere genet sett som har mange medlemmer som er under subtile endringer i uttrykk. Individuelt, disse endringene er ikke statistisk signifikante, men når en stor del av genene i et sett samlet skift i uttrykket, at trenden kan være svært signifikante statistisk sett.,
I tillegg til de mer sensitive algoritme, DAVIDS genet sett og gen-markeringer er egentlig gått ut på dato. Den siste oppdateringen av DAVIDS mange datakilder å være i midten til slutten av 2009. Som sådan, mange gen accessions fra Ensembl kanskje ikke støttes. Faktisk, fra min bakgrunn filen som inneholdt 19,030 bare 13,488 ble anerkjent av DAVID. Mangel på oppdatert genet sett er også en bekymring, som er den stadig voksende antall siteringer at DAVID fortsatt mottar.,
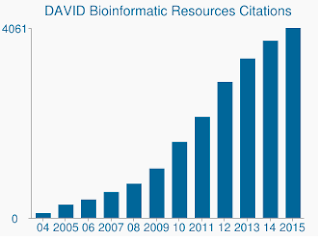
DAVID fortsatt mottar tusenvis av henvisninger per år til tross for underliggende data blir woefully ut på dato.
I konklusjonen, vil jeg anbefale forskere bruke verktøy som ser på en hel profil for å finne ut i vannmassene (GSEA, KAMERA, WilcoxGST, Pathifier), heller enn å sende genet sett for overlapper analyse. Ved hjelp av DAVID kan være den raske og enkle valget, men det kommer på bekostning av en massiv reduksjon i følsomheten.
Mer å lese
JA Timmons, KJ Szkop, IJ Gallagher., Flere kilder til skjevhet forvirre funksjonelle berikelse analyse av global-omics data. Genom biologi, 2015. 16:186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Virkningen av kunnskap opphopning på vei berikelse analyse. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: I Mai 2016, DAVID ble oppdatert!
https://david.ncifcrf.gov/content.jsp?file=release.html















