punctul genomului
analiza căii a devenit o procedură atât de comună în bioinformatică, în special în studierea expresiei genelor. Dacă te uiți la un sondaj de lucrări recente se pare că există o grămadă de moduri în care se poate face. În acest post, voi discuta diferențele dintre două instrumente utilizate în mod obișnuit; DAVID și GSEA.

conceptele Din spatele celor doi algoritmi sunt foarte diferite., DAVID determină suprapuneri între listele de gene furnizate de utilizator și bazele de date întreținute, căutând suprapuneri care sunt mai mari decât cele așteptate din întâmplare. Puteți îmbunătăți acuratețea algoritmului furnizând un fișier de fundal care conține toate genele care au fost considerate/detectate în experiment. Seturile de gene furnizate de utilizator sunt generate în mod normal prin selectarea genelor care trec un prag de semnificație. Procedura DAVID este similară cu altele disponibile, cum ar fi ingeniozitatea, AmiGO și GeneGO., Selectarea valorilor de semnificație este în mare măsură arbitrară, dar este comună setarea pragului la FDR ajustat p<0.05. Modificarea pragului va modifica considerabil rezultatele îmbogățirii. Testele statistice utilizate includ hipergeometric, Fisher exact și Chi-pătrat.
În contrast cu metodele de mai sus, care necesită liste de gene care trece praguri arbitrare, GSEA este un instrument care utilizează fiecare datapoint în algoritm statistic. În metoda” clasică ” genele sunt clasificate de la cele mai reglementate până la cele mai reglementate., Metrica rang în sine variază, dar două metode valide sunt de a utiliza semnat valoarea P, sau mai mici 90% interval de încredere de schimbare ori. Baza testului este de a evalua dacă membrii unui set de gene apar îmbogățiți la un capăt al profilului. Pentru a testa îmbogățirea, GSEA efectuează permutări ale profilului, calculând îmbogățirea setului de gene de o mie sau mai multe ori pentru a estima valorile p empiric. Alte instrumente care utilizează întregul profil efectuează teste analitice precum testul Wilcoxon și Kolmogorov-Smirnov.,
deci, acum că înțelegem principalele diferențe în algoritmi, putem începe să comparăm rezultatele acestor două tehnici. Am revizuit un set de date pe care l-am discutat înainte, tratamentul cu azacitidină în celulele AML3 profilate de ARN-seq. Voi folosi datele de Expresie diferențială ARN-seq generate de DESeq ca punct de plecare pentru a compara DAVID și GSEA.
pentru DAVID, am făcut un fișier de fundal format din toate cele 19.030 de gene detectate în experiment peste pragul de detectare. Valoarea semnificativă (FDR< 0.,05) listele de gene din direcția up (2502 gene) și down (2907 gene) au fost prezentate separat lui DAVID 6.7 pentru analiză, concentrându-se doar pe căile KEGG.
pentru GSEA, am folosit aceeași procedură metrică de rang descrisă anterior. Am folosit algoritmul clasic gsea cu gsea2-2.2.2.jar executabil, folosind Kegg seturi de gene de la MSigDB v5.1.
în graficul de mai jos, am determinat suprapunerea seturilor de gene semnificative statistic (FDR<0.05) separat pentru căile reglementate în sus și în jos., Am inclus, de asemenea, o intersecție a rezultatelor DAVID atunci când nu este furnizat niciun fundal (doar pentru curiozitate, nu este o procedură recomandată).
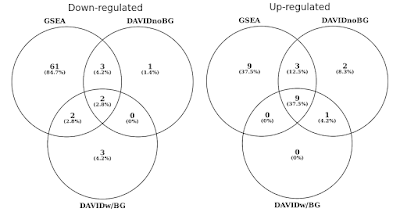
DAVID vs GSEA folosind același profil de expresie a genelor. Numerele reprezintă seturi de gene care sunt îmbogățite semnificativ (FDR<0,05).
veți observa că atât în direcția reglementată în sus, cât și în jos, GSEA identifică rezultate mai semnificative în comparație cu DAVID. Am constatat că acest lucru a fost cazul și în alte biblioteci de seturi de gene, cum ar fi REACTOME și GO., Diferența de sensibilitate dintre cele două instrumente se reduce la diferiți algoritmi, DAVID folosind o listă discretă de gene, față de GSEA care folosește fiecare punct de date. GSEA va fi mult mai sensibil în identificarea seturilor de gene care au mulți membri care suferă modificări subtile ale expresiei. În mod individual, aceste modificări nu sunt semnificative statistic, dar atunci când o mare parte a genelor dintr-un set colectiv își schimbă expresia, această tendință ar putea fi foarte semnificativă statistic vorbind.,
În plus față de algoritmul mai sensibil, seturile de gene ale lui DAVID și adnotările genelor sunt într-adevăr depășite. Ultima actualizare a numeroaselor surse de date ale lui DAVID fiind la mijlocul până la sfârșitul anului 2009. Ca atare, multe aderări genetice de la Ensembl nu pot fi acceptate. Într-adevăr, din dosarul meu de fond care conținea 19 030, doar 13 488 au fost recunoscute de DAVID. Lipsa seturilor de gene actualizate este, de asemenea, o preocupare, la fel ca și numărul tot mai mare de citări pe care DAVID le primește încă.,
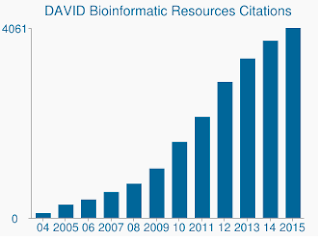
DAVID primește încă mii de citări pe an, în ciuda faptului că datele de bază sunt extrem de depășite.în concluzie, aș recomanda cercetătorilor să folosească instrumente care privesc un întreg profil pentru a determina îmbogățirea (GSEA, CAMERA, WilcoxGST, Pathifier), mai degrabă decât să prezinte seturi de gene pentru analiza suprapunerii. Utilizarea lui DAVID ar putea fi opțiunea rapidă și ușoară, dar aceasta vine în detrimentul unei reduceri masive a sensibilității.ja Timmons, kj Szkop, IJ Gallagher., Mai multe surse de părtinire confundă analiza de îmbogățire funcțională a datelor global-omics. Genome biology, 2015. 16: 186
Lina Wadi, Mona Meyer, Joel Weiser, Lincoln D Stein, Juri Reimand. Impactul acumulării de cunoștințe asupra analizei de îmbogățire a căii. bioRxiv. doi: http://dx.doi.org/10.1101/049288
EDIT: în Mai 2016, DAVID a fost actualizat!
https://david.ncifcrf.gov/content.jsp?file=release.html















